Imagine you are in a lab, and the experiment you are about to run depends on a simple act of trust. You open a supplier’s catalog, choose an antibody, and look at a validation image called a Western blot. For a non-specialist, a Western blot is basically a lab test that shows whether an antibody recognizes the right protein: if the right dark band appears in the right place, scientists take it as evidence that the antibody works.
Now imagine someone zooming in, adjusting the contrast, and discovering that some of those bands may have been copied, flipped, painted over, or reused across different product pages. Suddenly, the problem is no longer one questionable image. It is a much larger question: how much of the scientific supply chain are we trusting without truly verifying?
A recent Nature article reported that catalog entries for more than 100 Thermo Fisher Scientific antibodies contained images that appeared to have been manipulated — including images intended to demonstrate antibody quality and performance . Image alteration does not automatically mean the underlying products are defective, but the episode is a stark reminder that even trusted commercial data streams can carry hidden uncertainty.
What makes the Thermo Fisher case troubling is that the reported problems were not limited to a single figure or one isolated product page. Reese Richardson’s analysis identified “more than 100 images bearing signs of manipulation” in the online primary-antibody catalog: Western blot bands that appear identical after flipping and rotation, conspicuous brushstroke-like edits after contrast adjustment, repetitive blocks of background noise, and abrupt discontinuities in background texture. In one recurring pattern, dozens of pages showed verification blots sharing the same background, with 50 instances documented at the time of writing.
A principal investigator facing allegations of duplicated bands, rotated blot features, and repeated backgrounds across many figures would expect institutional investigation, retractions, grant scrutiny, and possibly the collapse of their lab’s credibility. So the uncomfortable question for industry is this: if comparable problems can appear in commercial verification data that thousands of scientists rely on, what does that mean for every small biotech, CRO, platform company, and pharma organization whose reagent choices, model-training pipelines, and therapeutic programs may depend on that data?
None of this is new. Nature’s broader coverage shows that reproducibility is a structural issue, not a one-off scandal. In its 2016 survey, two-thirds of responding researchers viewed current levels of reproducibility as a major problem, with pressure to publish, selective reporting, poor statistics, and finicky protocols all contributing (Nature).
The issue is especially acute in preclinical biology. Nature Reviews Drug Discovery reported that Bayer could replicate only about 25% of the preclinical academic projects it took on, while Amgen reported an 11% success rate when trying to recreate findings from cancer papers (Nature Reviews Drug Discovery). Those numbers should matter to every company building computational platforms for drug discovery, because they carry a very practical consequence:
Pharma will not simply trust the model output because the model looks sophisticated.

If large companies have already struggled to reproduce a major fraction of published preclinical biology, any AI-first company selling predictions, targets, or mechanisms should expect tougher diligence — more requests for raw data, assay details, reagent provenance, replication, and evidence that the model is learning real biology rather than artifacts. In practice, weak trust in data reproducibility lengthens every partnering cycle.
This is also why external data alone is not enough. CROs are essential partners, but outsourced data often arrives detached from the tacit experimental context that makes biology interpretable: reagent history, protocol drift, failed runs, operator effects, batch artifacts, and subtle assay behaviors. Those details often decide whether a model learns biology or learns noise. A model trained on weakly controlled data can become very good at reproducing hidden artifacts — confident in silico, and wrong in the real world. The lesson I keep coming back to is that trust in data has to be earned experimentally, repeatedly, and transparently, by owning the critical feedback loops yourself.
But there is a second, subtler trap — and a paper published this week brought it into sharp focus for me.
There is a comforting story we have all imported from language and vision models: more data, bigger model, better performance. Single-cell biology borrowed that playbook wholesale, scaling foundation models from corpora of one million cells to atlases of more than a hundred million, on the assumption that scale alone would unlock the same gains.

A new study in Nature Methods by Alan DenAdel and colleagues put that assumption to a brutal test (DenAdel et al., Nature Methods, 2026). Working from a 22.2-million-cell corpus, they pretrained 400 models across five architectures — from humble PCA and a variational autoencoder up to the Geneformer transformer — and ran 6,400 evaluation experiments. As Jorge Bravo-Abad summarised it, the result is sobering (@bravo_abad).
Performance saturated almost immediately. On cell-type classification, batch integration, and perturbation prediction, most models hit their ceiling at roughly 1% of the corpus — about 200,000 cells.
Beyond that, adding millions more cells changed essentially nothing (@bravo_abad). More diversity did not help.
Even spiking in genome-scale Perturb-seq data — feeding the models perturbed phenotypes rather than just healthy cells — failed to move the needle (DenAdel et al.).
Two findings hit hardest.
First, simple baselines like PCA and logistic regression often matched or beat the transformers (@bravo_abad).
Second, the strongest model won not because it was bigger, but because its training objective was aligned with the actual downstream task.
In other words, what you train on and how you frame the question matter far more than how much you collect.
In a field already wrestling with reproducibility, is throwing the kitchen sink at biological data generation actually a good idea — when massive cellular datasets show so little tangible benefit?
I do not think it is. The instinct to keep scaling pretraining corpora may be burning enormous compute for no return. The real leverage sits somewhere far less glamorous: curating high-quality, task-relevant data, and matching the model to the question you are genuinely trying to answer.
If the real leverage lies in matching effort to the question that actually matters, then it is worth asking where the hardest, highest-value questions in human biology live. For me, the answer has always been the same: in the proteins that refuse to hold still.
We were taught a tidy story — sequence folds into structure, structure dictates function. But roughly a third of the residues in the human proteome never fold into a fixed shape at all, and nearly 60% of human proteins contain at least one such intrinsically disordered region
Source: Pritišanac et al., “A Functional Map of the Human Intrinsically Disordered Proteome,” bioRxiv2024.
These intrinsically disordered proteins and regions (IDPs/IDRs) are not broken or leftover; they exist as shifting ensembles of interconverting conformations, and that very flexibility is what lets them do their jobs — a principle now formalized as the “disorder–function paradigm” (Trivedi & Nagarajaram, Int. J. Mol. Sci. 2022).
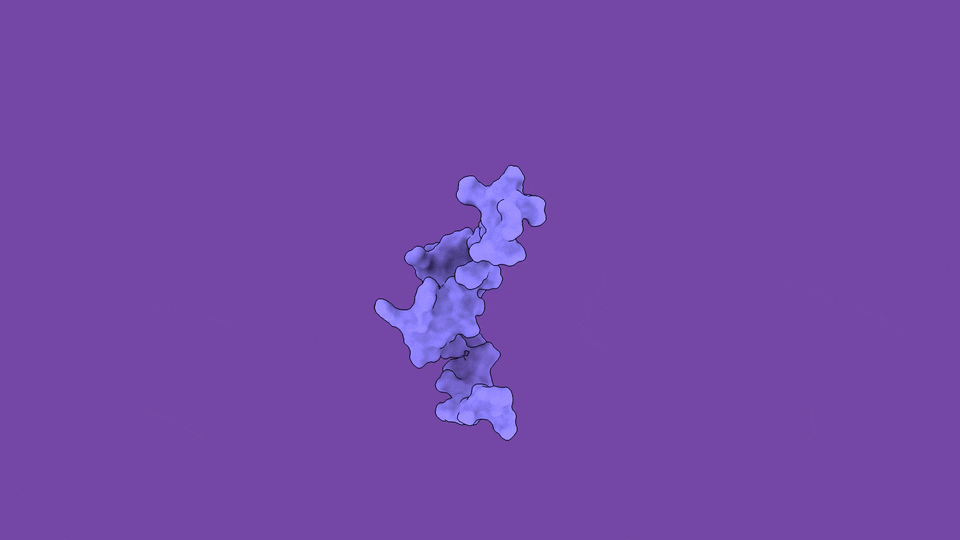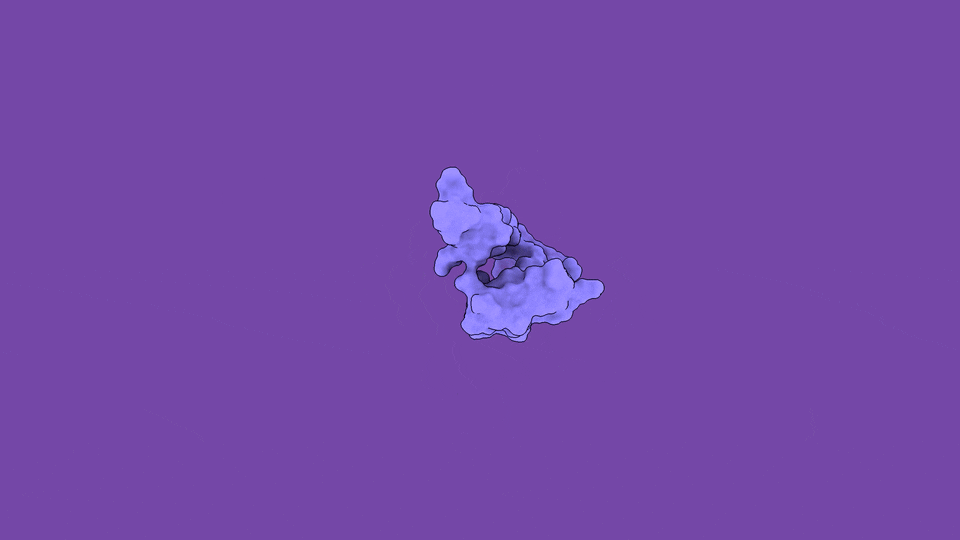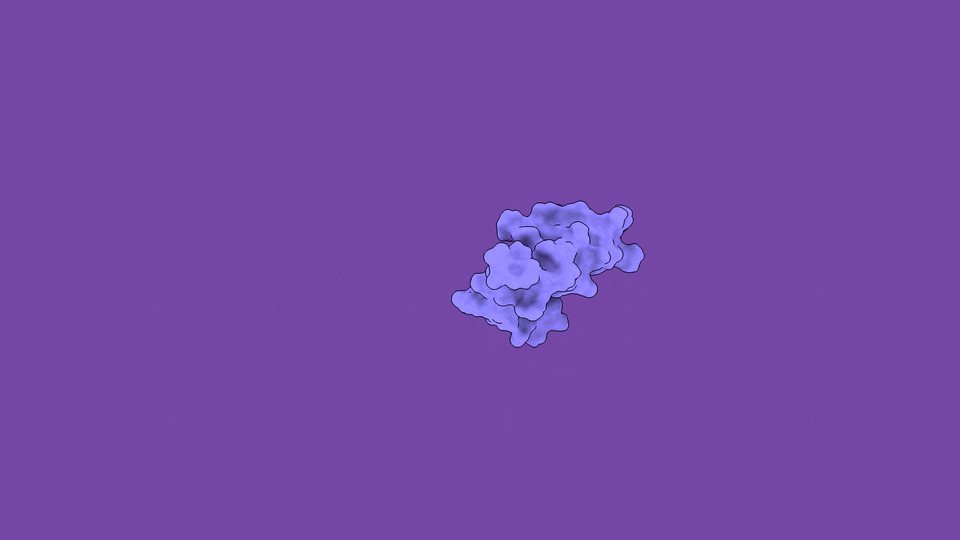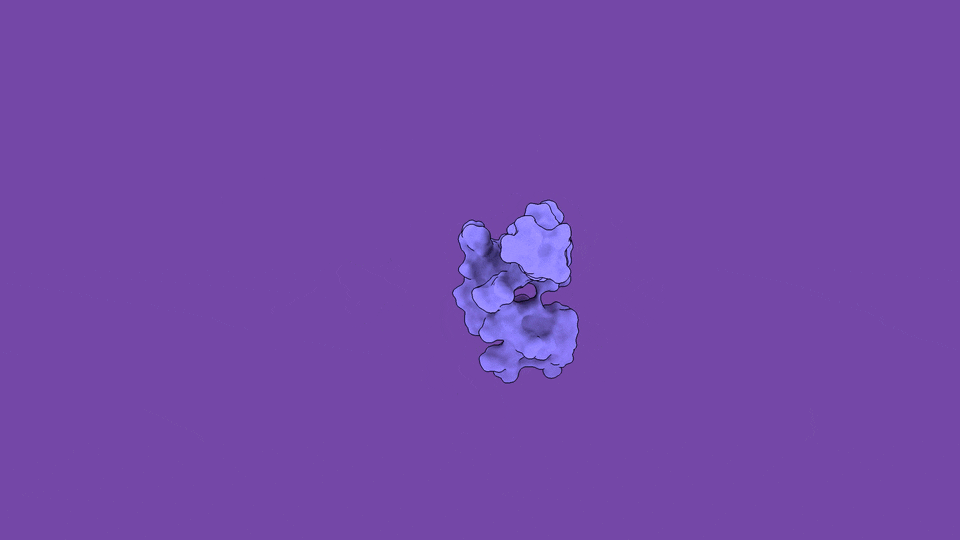
The jobs they do are not peripheral. Disordered regions sit at the controlling hubs of biology — transcription, cell signaling, and the formation of the membraneless condensates that organize the inside of a cell — which is precisely why their dysregulation is so often catastrophic (Babu et al., Curr. Opin. Struct. Biol. 2011). When you trace the molecular roots of our most stubborn diseases, you keep arriving at disordered proteins:
Cancer. The master tumor suppressor p53 and a long list of oncogenic signaling proteins are rich in disorder, and IDRs are frequently the site of disease-associated mutations (Uversky et al., Chemical Reviews 2014).
Neurodegeneration. Amyloid-β, tau, and α-synuclein — the disordered proteins at the heart of Alzheimer’s, Parkinson’s, Huntington’s, and ALS — misfold and aggregate in ways that drive the disease (“Intrinsically disordered proteins and proteins with intrinsically disordered regions in neurodegenerative diseases,” Biophysical Reviews 2022).
Disorder is not an exotic edge case. It is where a huge fraction of disease biology actually happens — and where our tools have been weakest.
And here is the painful irony. These same proteins have, for decades, carried the label “undruggable.” More than half of the human proteome is too flexible to present the neat, static pocket that classical, structure-based drug discovery was built to exploit, so the field largely walked past it (Xie et al., Signal Transduction and Targeted Therapy 2023).
The targets with the most therapeutic upside have been the very ones we understood — and could measure — the least. That gap is not a reason to look away. It is the single most compelling scientific opportunity I know.
These are precisely the questions in fundamental structural biology that have fascinated my team and me for years — long before “foundation model” was a phrase anyone said out loud. How does a protein that has no fixed structure still do something exquisitely specific? Where does the signal actually live? And what kind of data do you need to capture it?
That fascination is why we embarked on the journey to build Peptone: a company devoted to proteins, where experimental rigour and deep expert knowledge are not support functions but the gate to scientific discovery. We did not set out to collect the most data. We set out to generate the right data, for the right targets, interrogated by people who understand the physics underneath.
And nowhere is that approach more necessary than with the hardest targets of all: intrinsically disordered proteins.
Our latest paper in Nature Communications makes this concrete (Streit, Invernizzi, Bottaro, Tamiola & Lindorff-Larsen, 2026). Roughly a third of all protein residues in our cells are intrinsically disordered — they do not fold into a single, tidy structure, but populate broad, shifting ensembles. They drive everything from transcription to signaling, and they are notoriously difficult to characterize, whether you reach for an experiment or a computer.

In this work, we applied an established enhanced-sampling approach — On-the-fly Probability Enhanced Sampling (OPES) in a multithermal ensemble — to disordered proteins, showing that a single simulation can efficiently explore their conformational landscape without the brittle parameter tuning and replica-exchange machinery these simulations normally demand.
The headline finding is the part that still gives me chills. We applied the method to ACTR, a 71-residue disordered transcriptional coactivator that folds only when it meets its partner. In its free state, ACTR looks like a floppy, structureless chain. But our simulations revealed something experiments alone cannot see: a rare, low-population (~3%) set of transiently structured states in which multiple α-helices fold cooperatively and form genuine tertiary contacts — a fleeting, binding-competent shape that the protein visits and abandons, over and over.
These hidden states are not a numerical artifact. They are reversibly sampled, separated from the disordered ground state by a modest free-energy barrier of only ~13 kJ/mol, and — critically — consistent with extensive NMR and SAXS data we used to validate the ensemble (Nature Communications). The physics and the experiments agree. And the payoff is that these transient, partially folded conformations may harbor exactly the kind of binding pockets that make an “undruggable” disordered protein suddenly addressable.
You do not find these hidden states by collecting more data. You find them by running the right physics in computers, interpreted by people who know what they are looking at.
This is the whole thesis behind Peptone. And it begins with a simple, often-overlooked fact: the data we generate simply does not exist in the public domain. There is no atlas to download, no benchmark corpus to fine-tune on, no repository quietly holding the answers for the disordered targets we care about. Disordered proteins have been systematically left out of the structural record precisely because they refuse to sit still for the classical tools.
So if you want this data, there is only one option — you have to make it yourself.
That is exactly what we do. We combine bespoke physical-reality simulations with deep expert knowledge and highly specialized experimental data — data that is always generated for the specific disordered target we are working on, never pulled off a shelf or borrowed from a generic atlas.
A large part of our unfair advantage lies in how we generate that data. Our hydrogen–deuterium exchange mass spectrometry (HDX-MS) capability is prototypical — built and tuned in-house specifically to interrogate disordered proteins, not adapted from an off-the-shelf workflow designed for well-folded ones. HDX-MS lets us read, residue by residue, how exposed or protected each part of a protein chain is, which means we can capture the fleeting, partially structured states that define disorder and confirm whether the conformations our simulations predict are real. Because almost no one else is generating this kind of data on these targets, every measurement compounds into an advantage that cannot simply be bought or scraped: it has to be earned, experiment by experiment.
The result is technology that models disordered proteins of any size or complexity and reveals the “invisible” pockets that conventional structure-based drug discovery walks straight past (Peptone).

It is the exact inverse of the kitchen-sink approach. Smaller, more specialized models. Data we understand because we made it. Physics done well, blended with empirical measurement and human expertise.
The tangible fruits of this expert approach are not abstract:
Exponentially accelerated drug development. When your model learns real biophysics instead of artifacts, every cycle of design, measurement, and decision gets tighter and faster.
Genuinely novel biological insight. Transient hidden states, cooperative folding events, cryptic pockets — discoveries that simply are not visible to experiment or generic AI alone.
A real shortening of the time to the clinic. Fewer dead ends, fewer irreproducible surprises, and more confidence that what we see in silico will hold up in the lab and the patient.
The future of computational drug discovery is not “more data instead of labs,” and it is not “the biggest model wins.” It is smaller, smarter, bespoke — models built on data we generate, understand, and trust, aimed squarely at the most difficult and most exciting targets in human biology.
That is why, for us, bespoke data generation is not a feature.
It is the foundation.
If you want to learn more about what we are building at Peptone, please see this short flick put together with NVIDIA and AWS.