
Part I: The AlphaFold Revolution
By OpenMed, Open-Source Agentic AI for Healthcare & Life Sciences
TL;DR: The 2024 Nobel Prize in Chemistry went to the creators of AlphaFold, a deep learning system that solved a 50-year grand challenge in biology. The architectures behind it (transformers, diffusion models, GNNs) are the same ones you already use. This post maps the protein AI landscape: key architectures, the open-source ecosystem (which has exploded since 2024), and practical tool selection. Part II (coming soon) covers how I built my own end-to-end pipeline.
Table of Contents
- Introduction: The Biggest ML Story You Might Have Missed
- Biology Foundations for ML People
- AlphaFold: The Architecture Revolution
- The Open-Source Ecosystem
- Tool Selection Guide
- The Path Forward
- Key References
1. Introduction: The Biggest ML Story You Might Have Missed
On October 9, 2024, the Nobel Prize in Chemistry went to Demis Hassabis and John Jumper of Google DeepMind for AlphaFold, alongside David Baker for computational protein design. First time a Nobel in Chemistry went primarily to machine learning researchers.
AlphaFold 2 was published in 2021. Three years from conference paper to Nobel Prize. That timeline reflects how transformative this work has been.
The techniques that power AlphaFold aren't exotic biology tools. They're transformers, attention mechanisms, diffusion models, and graph neural networks. Protein folding has become one of the most active frontiers for architectural innovation in deep learning.
ML Concepts Meet Biology
| ML Concept | Protein Application | Why It's Interesting |
|---|---|---|
| Transformers & Attention | Evoformer uses novel axial and triangle attention | Attention patterns designed for 2D relationship matrices |
| Diffusion Models (DDPM) | AlphaFold 3, RFdiffusion generate 3D structures | Denoising in SE(3) space with physical constraints |
| Graph Neural Networks | ProteinMPNN treats proteins as geometric graphs | Message passing on 3D point clouds |
| Language Models (MLM) | ESM-2 learns protein "grammar" | Masked prediction reveals evolutionary patterns |
| SE(3) Equivariant Networks | Structure modules preserve 3D symmetry | Outputs unchanged by rotation/translation of inputs |
| Multi-Modal Learning | Chai-1, Boltz combine sequence + structure + ligands | Fusing heterogeneous biological data |
| Generative Models | ESM-3, Chai-2 generate novel proteins and antibodies | Protein design, not just prediction |
The Stakes Are High
This matters outside of ML benchmarks. Accurate structure prediction is already reshaping:
- Drug Discovery: From years to weeks for lead identification
- Vaccine Development: Rapid antigen design (as we saw with COVID-19)
- Enzyme Engineering: Custom catalysts for industrial processes
- Gene Therapy: Optimized delivery vectors
And most of the best tools are open-source.
2. Biology Foundations for ML People
You don't need a biology degree to work with protein AI, but you do need a few key concepts.
Proteins: The 20-Letter Language
Proteins are the molecular machines that power all of life, sequences written in a 20-letter alphabet of amino acids. A typical protein is 100 to 1,000 amino acids long.
What proteins do:
- Enzymes (like amylase) break down molecules (starch into sugar in your saliva)
- Antibodies recognize and neutralize viruses and bacteria
- Hemoglobin carries oxygen through your bloodstream
- Insulin regulates blood sugar levels
- Collagen provides structural support to skin and bones
A protein's function is determined by its 3D shape. The same sequence always folds into the same structure, and that structure determines what the protein can do. Understanding shape = understanding function.
The Central Dogma: DNA → RNA → Protein
To manufacture a protein, cells follow a two-step process:
DNA (gene) → [Transcription] → mRNA → [Translation] → Protein
- Transcription: The DNA gene is copied into a messenger RNA (mRNA) molecule
- Translation: The ribosome reads the mRNA and assembles the amino acid chain
The genetic code uses codons (three-letter sequences) to specify each amino acid:
ATG→ Methionine (start signal)TGA,TAA,TAG→ Stop signals- Most amino acids have 2 to 6 different codons (redundancy)
This redundancy becomes important for mRNA optimization, which I cover in Part II.
The Protein Folding Problem
Input: A sequence of amino acids (like MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSH...)
Output: 3D coordinates for every atom in the protein
Challenge: The search space is impossibly large, which brings us to Levinthal.
Levinthal's Paradox: Why Brute Force Fails
Cyrus Levinthal calculated that if you tried every possible protein conformation at 10¹² configurations per second, you'd need longer than the age of the universe to find the right one for a single small protein.
Yet nature folds proteins in milliseconds.
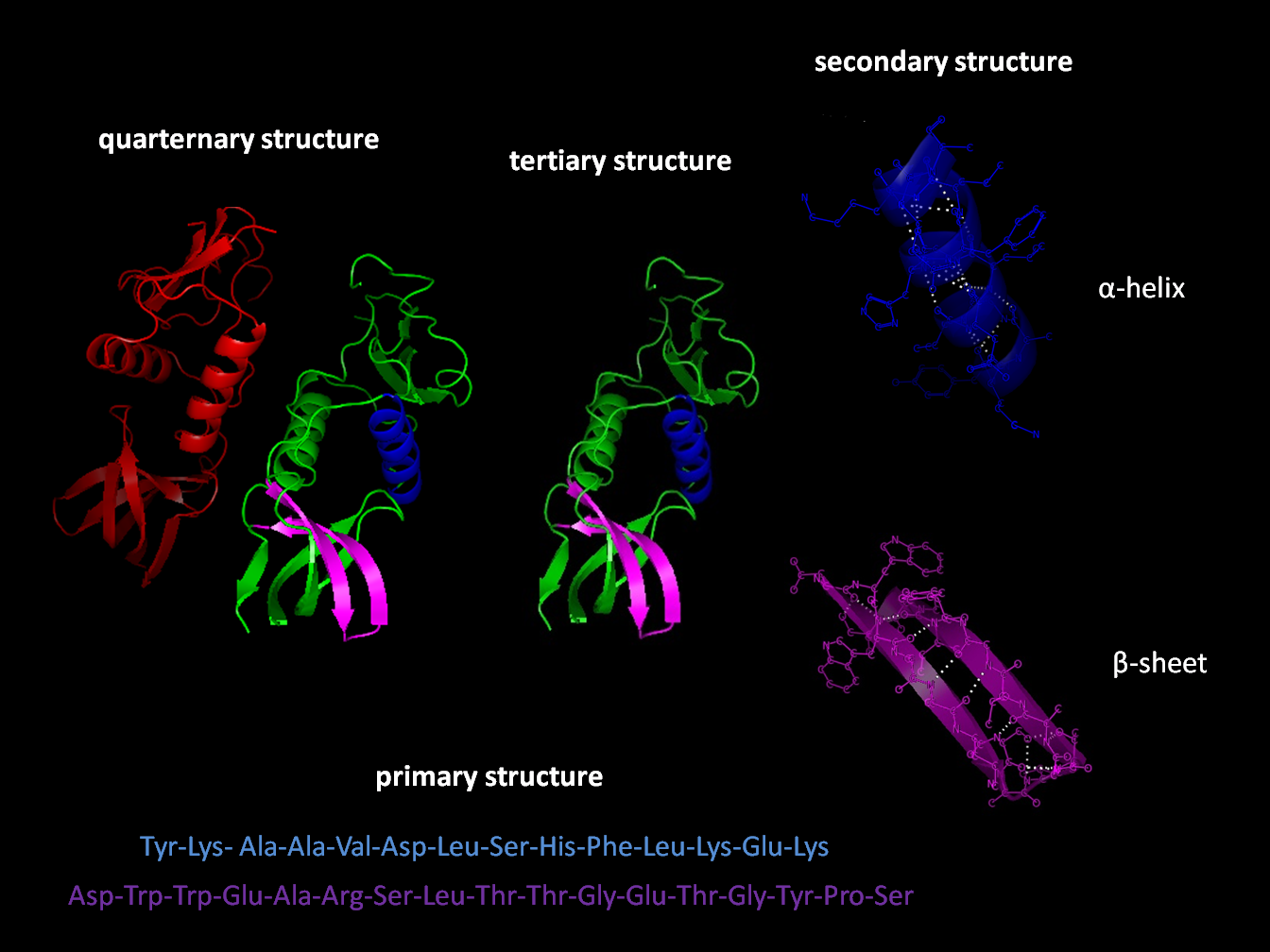 Figure: Protein folding levels. Source: Wikipedia
Figure: Protein folding levels. Source: Wikipedia
The Key Insight: Co-evolution Encodes Structure
When two positions in a protein need to be physically close in 3D space, they co-evolve together across species. If position 5 mutates, position 50 (which touches it in 3D) often mutates in a compensating way to maintain the interaction.
By analyzing millions of related sequences across species (called Multiple Sequence Alignments or MSAs), you can infer which positions interact, and therefore what the 3D structure looks like. It's the same idea as learning word embeddings from co-occurrence patterns, except instead of words co-occurring in sentences, amino acids co-evolve across billions of years. The "corpus" is the tree of life itself.
3. AlphaFold: The Architecture Revolution
In November 2020, DeepMind entered CASP14 (a biennial competition where teams predict protein structures from sequence) and AlphaFold 2 achieved a median GDT score of 92.4. For context, above 90 is considered comparable to experimental methods. AlphaFold didn't just win; it essentially solved the competition.
AlphaFold 2: The Breakthrough
The High-Level Architecture
Input → Evoformer (48 blocks) → Structure Module → Output
┌─────────────────┐ ┌──────────────────────┐ ┌──────────────────┐ ┌─────────┐
│ MSA (N×L×feat) │ ──── │ Row Attention │ ─── │ IPA (Invariant │ ── │ 3D xyz │
│ Pair (L×L×feat) │ │ Column Attention │ │ Point Attention) │ │ coords │
│ Templates │ │ Triangle Updates │ │ Angle Prediction │ │ │
└─────────────────┘ └──────────────────────┘ └──────────────────┘ └─────────┘
 Figure: AlphaFold 2 architecture and CASP14 performance. Panel (e) shows the full pipeline: input features → Evoformer → Structure Module → 3D coordinates. Source: Jumper et al., Nature 2021 (CC BY 4.0)
Figure: AlphaFold 2 architecture and CASP14 performance. Panel (e) shows the full pipeline: input features → Evoformer → Structure Module → 3D coordinates. Source: Jumper et al., Nature 2021 (CC BY 4.0)
Key Innovations
1. The Evoformer Block
The Evoformer is a novel transformer variant that processes two representations simultaneously:
MSA Representation (N sequences × L positions): A batch of related sequences. Row attention lets each position learn from the same position across different species. Column attention lets each sequence learn from neighboring positions.
Pair Representation (L × L matrix): Encodes the relationship between every pair of positions. Like a graph attention network, but with a dense learned representation instead of a sparse adjacency matrix.
The Evoformer is essentially a Vision Transformer that jointly processes an image (the MSA as a 2D grid) and a graph (the pair representation), with bidirectional information flow between them.
2. Triangle Updates: Enforcing Geometric Consistency
Triangle updates enforce geometric consistency: if position A is close to B, and B is close to C, then the A-C relationship must be consistent. They update the pair representation using:
- Triangle multiplication (outgoing): Aggregate information about A-C via all intermediate positions B
- Triangle multiplication (incoming): The reverse direction
- Triangle attention: Self-attention with triangle-structured masks
This is transitivity enforcement for a graph, similar to path aggregation in GNNs, but formulated as attention operations. The network learns that 3D space has geometric constraints: you can't have A close to B, B close to C, but A far from C.
3. Invariant Point Attention (IPA)
The structure module uses SE(3)-equivariant attention. This is attention that respects 3D geometry:
- Each residue has a "frame" (position + orientation in 3D)
- Attention scores are computed using both sequence features AND 3D distances
- The output is guaranteed to transform correctly if you rotate or translate the input
If you've worked with equivariant networks (E(n)-GNNs, SE(3)-Transformers), this is that family. Rotate the input protein by 90°, the output rotates by exactly 90°. No data augmentation needed. The symmetry is baked into the architecture.
4. Recycling: Iterative Refinement
AlphaFold doesn't predict the structure in one shot. It runs the network 3 times, feeding the output of each iteration back as input to the next. Each pass refines the prediction.
Similar to iterative refinement in diffusion models or flow matching, but without explicit noise. Also reminiscent of iterative amortized inference in VAEs.
The MSA Bottleneck
One major practical issue: MSA generation is slow.
For each protein, AlphaFold must:
- Search massive sequence databases (UniRef, MGnify, BFD)
- Run HHblits/JackHMMer to build alignments
- This takes minutes to hours per protein
The model inference itself is fast (minutes on GPU). But the MSA search makes high-throughput applications impractical with vanilla AlphaFold 2.
AlphaFold 3 (2024): Enter Diffusion
In 2024, DeepMind released AlphaFold 3 with a major architectural shift: diffusion-based structure generation.
What Changed
| Aspect | AlphaFold 2 | AlphaFold 3 |
|---|---|---|
| Scope | Proteins only | Proteins + DNA + RNA + ligands + ions |
| Architecture | Evoformer + IPA | Pairformer + Diffusion |
| Structure Prediction | Direct coordinate regression | Diffusion-based denoising |
| Output | Single structure | Ensemble of structures |
| License | Apache 2.0 ✅ | Non-commercial ❌ |
 Figure: AlphaFold 3 pipeline and performance across biomolecular complex types. Panel (d) shows the inference architecture: Pairformer trunk → diffusion module → 3D atomic coordinates. Source: Abramson et al., Nature 2024 (CC BY 4.0)
Figure: AlphaFold 3 pipeline and performance across biomolecular complex types. Panel (d) shows the inference architecture: Pairformer trunk → diffusion module → 3D atomic coordinates. Source: Abramson et al., Nature 2024 (CC BY 4.0)
The Diffusion Module
AF3 uses a Denoising Diffusion Probabilistic Model (DDPM) for structure prediction:
- Start with noisy atomic coordinates (Gaussian noise)
- Iteratively denoise using a learned score function
- The denoiser is conditioned on the Pairformer embeddings
- Multiple samples give uncertainty estimates
It's image diffusion, but the "image" is a 3D point cloud. The denoiser is SE(3)-equivariant, and the noise schedule and architecture are adapted for molecular coordinates rather than pixels.
The Licensing Situation
DeepMind released AF3's code and weights in November 2024, a significant move. But the license remains non-commercial. If you're building a commercial application, you can't use AF3 directly. The community response has been remarkable though: multiple open-source reproductions now match or exceed AF3's accuracy with permissive licenses (more below).
4. The Open-Source Ecosystem
The AlphaFold breakthrough sparked an explosion of open-source development. Today, you don't need to use DeepMind's code. There's a rich ecosystem of alternatives, each with different strengths.
The Official DeepMind Tools
The AlphaFold Database is particularly valuable. It contains predictions for nearly every known protein, so check there before running your own.
Structure Prediction: The Open Alternatives
OpenFold: AF2 in PyTorch
If you're a PyTorch person (and most ML engineers are), OpenFold is your entry point. It's a faithful reimplementation of AlphaFold 2 that matches the original's accuracy (GDT-TS correlation > 0.99).
Why it matters:
- Trainable: Unlike DeepMind's JAX code, you can actually fine-tune it
- Familiar: Standard PyTorch, integrates with your existing workflows
- Well-documented: Active community, good tutorials
Links: GitHub | Paper (Nature Methods 2024)
ESMFold: The Language Model Approach
ESMFold from Meta AI skips MSA generation entirely. Train a massive language model (ESM-2, up to 15B parameters) on 65 million protein sequences using masked language modeling. The model learns evolutionary patterns implicitly from sequence context alone. Add a folding head, and you get 3D coordinates from a single sequence in seconds.
| Model | Accuracy (TM-score) | Speed | MSA Required |
|---|---|---|---|
| AlphaFold 2 | 0.92 | Hours | Yes |
| ESMFold | 0.87 | Seconds | No |
| OmegaFold | 0.85 | Seconds | No |
ESMFold is to AlphaFold what GPT is to retrieval-augmented systems. Instead of explicitly retrieving related sequences (MSA), it has internalized the patterns during pre-training.
When to use ESMFold:
- High-throughput screening (millions of proteins)
- "Orphan" proteins with no known relatives
- Real-time applications
- Limited compute budget
When to use AlphaFold 2 instead:
- Maximum accuracy is critical
- You need confident domain boundaries
Links: GitHub | HuggingFace | Paper (Science 2023)
ESM-3 (EvolutionaryScale, June 2024; published in Science January 2025) is the next generation, a multimodal generative model that operates across sequence, structure, and function simultaneously. It generated a novel GFP (green fluorescent protein) with only 58% sequence identity to known fluorescent proteins, demonstrating genuine generative capability. This is protein generation, not just prediction.
ESM-C (December 2024) is a drop-in replacement for ESM-2 in embedding workflows. The 300M parameter model matches ESM-2 650M performance. Same API, half the compute.
Links: ESM-3 Paper (Science 2025) | EvolutionaryScale
The AF3 Alternatives (and Beyond)
The AF3 non-commercial license created a gap, and the community filled it fast. Multiple open-source models now match AF3's capabilities, and the latest generation goes well beyond structure prediction.
| Tool | Capabilities | License | Link |
|---|---|---|---|
| Chai-1 | Proteins + ligands + DNA/RNA | Apache 2.0 ✅ | GitHub |
| Chai-2 (Jun 2025) | Generative antibody design, 16% hit rate in de novo design (>100x over prior methods) | Apache 2.0 ✅ | GitHub |
| Boltz-1 | Biomolecular interactions | MIT ✅ | GitHub |
| Boltz-2 (Jun 2025) | Structure + binding affinity prediction. Approaches physics-based FEP accuracy at 1000x less compute | MIT ✅ | GitHub |
| Protenix (ByteDance, Feb 2026) | PyTorch AF3 reproduction. Protenix-v1 outperforms AF3 | Apache 2.0 ✅ | GitHub |
| OpenFold3 (Oct 2025) | AF3 reproduction from 30+ organizations | Apache 2.0 ✅ | GitHub |
| RF-AA | All-atom (Baker Lab) | BSD-3 ✅ | GitHub |
Boltz-2 is the first model to predict both structure and binding affinity in a single forward pass. For drug discovery, this is a big deal. Binding affinity estimation traditionally requires expensive physics-based free energy perturbation (FEP) calculations. Boltz-2 approaches that accuracy at a fraction of the compute.
Chai-2 moved beyond structure prediction into generative antibody design. A 16% hit rate in de novo antibody design may not sound high, but the previous best methods were under 0.1%. That's more than 100x improvement.
Protenix from ByteDance is a clean PyTorch AF3 reproduction with Apache 2.0 licensing (commercially friendly, unlike AF3 itself). Their v1 release actually outperforms the original AF3 on standard benchmarks.
OpenFold3 from the OpenFold Consortium brings the same open-source ethos that made OpenFold (the AF2 reproduction) so valuable. Over 30 organizations contributed.
ColabFold: The Practical Choice
ColabFold deserves special mention. It makes AlphaFold 2 actually usable by:
- Replacing the slow MSA search with MMseqs2 (100x faster)
- Providing Google Colab notebooks (free GPU!)
- Supporting batch processing
The result: 10-100x faster than vanilla AF2 with the same accuracy. This is what most researchers actually use day-to-day.
Links: GitHub | Paper (Nature Methods 2022)
Protein Design: The Inverse Problem
Structure prediction goes sequence → structure. Protein design goes the other direction: given a target 3D shape, what sequence will fold into it? This is inverse folding, and it's essential for engineering new proteins.
ProteinMPNN: The Gold Standard
ProteinMPNN from the Baker Lab treats proteins as geometric graphs:
- Nodes: Amino acid residues
- Edges: Spatial proximity (K-nearest neighbors in 3D)
- Message passing: Information flows between spatially adjacent residues
Structure Graph → Encoder (GNN) → Sequence Decoder (Autoregressive) → Amino Acid Sequence
The model generates sequences autoregressively. Each amino acid is predicted based on the structure AND all previously predicted amino acids.
Performance:
- 47% native sequence recovery (recovering nearly half the original amino acids from structure alone)
- >50% experimental success rate in wet-lab validation
- ~1 second per design
Architecturally, it's a GNN encoder with an autoregressive decoder, similar to graph-to-sequence models in NLP, but the graph is defined by 3D spatial proximity rather than explicit edges.
Links: GitHub | Paper (Science 2022)
RFdiffusion: De Novo Design
 Figure: Protein design using RFdiffusion. Panel (a) shows the denoising trajectory from random noise (t=T) to a folded protein backbone (t=0). Source: Watson et al., Nature 2023 (CC BY 4.0)
Figure: Protein design using RFdiffusion. Panel (a) shows the denoising trajectory from random noise (t=T) to a folded protein backbone (t=0). Source: Watson et al., Nature 2023 (CC BY 4.0)
RFdiffusion generates entirely new protein structures using SE(3)-equivariant diffusion:
- Start from noise
- Iteratively denoise to produce a new fold
- Can be conditioned on functional motifs ("design around this binding site")
Generative AI for protein structures, and it works. Designs have been validated experimentally.
RFdiffusion3 (November 2025) is a complete rewrite: atom-level precision, 10x faster, and handles protein-DNA, small molecule, and enzyme design. Training code released.
Links: RFdiffusion GitHub | Paper (Nature 2023) | RFdiffusion3 GitHub
Other Design Tools
| Tool | Approach | Advantage |
|---|---|---|
| LM-Design | Language model + structural adapters | 55-57% recovery (SOTA) |
| PiFold | Non-autoregressive | 70x faster than ProteinMPNN |
Production & Scale
For high-throughput work, you'll need infrastructure:
| Tool | Purpose | Link |
|---|---|---|
| MMseqs2 | Fast sequence search (400x faster than BLAST) | GitHub |
| AlphaPulldown | Screen protein-protein interactions | GitHub |
| AF2Complex | Reuse features for complex prediction | GitHub |
Training Data
If you want to train your own models:
| Resource | What It Contains | Link |
|---|---|---|
| OpenProteinSet | MSAs for ~140K protein families | GitHub |
| PDB | ~220K experimental structures | rcsb.org |
| UniProt/UniRef | Protein sequence databases | uniprot.org |
5. Tool Selection Guide
The ecosystem is big enough now that picking the right tool is a real decision. My rough heuristic: start with what your task actually requires. If you just need a structure, ColabFold is still hard to beat. If you need protein-ligand interactions, you're choosing between Chai-1, Boltz-1, and Protenix, all AF3-class, all commercially usable. If you need to design something, the Baker Lab tools (ProteinMPNN, RFdiffusion3) remain the gold standard.
By Task
| What You Need | Recommended Tool | Why |
|---|---|---|
| Single protein, max accuracy | ColabFold / AlphaFold 2 | Gold standard, MSA-based |
| Single protein, fast | ESMFold | Seconds, no MSA |
| Protein + small molecule | Chai-1, Boltz-1, or Protenix | AF3-level, commercial-friendly |
| Protein + DNA/RNA | RF-AA or RFdiffusion3 | Handles nucleic acid complexes |
| Protein complex | AlphaFold-Multimer | Multi-chain predictions |
| Binding affinity prediction | Boltz-2 | Structure + affinity in one pass |
| High-throughput (millions) | ESMFold | Speed at scale |
| Protein embeddings | ESM-C (non-commercial) | Drop-in ESM-2 replacement, faster |
| Design: structure → sequence | ProteinMPNN | Battle-tested, high success rate |
| Design: generate new structure | RFdiffusion3 | Atom-level, 10x faster than v1 |
| Design: de novo antibodies | Chai-2 | 16% hit rate, >100x improvement |
| Protein generation (multimodal) | ESM-3 (non-commercial) | Sequence + structure + function |
| Train custom models | OpenFold + OpenProteinSet | Full pipeline available |
Licensing deserves its own table because it's a real constraint. Some of the best models (AF3, ESM-3, ESM-C) are non-commercial. If you're at a startup or building a product, this narrows your options, but the commercial-friendly ecosystem is strong enough that you're not missing much.
By License (For Commercial Use)
| Tool | License | Commercial OK? |
|---|---|---|
| ESMFold / ESM-2 | MIT | ✅ Yes |
| ESM-3 | Cambrian (non-commercial) | ❌ No |
| ESM-C | Cambrian (non-commercial) | ❌ No |
| ProteinMPNN | MIT | ✅ Yes |
| OpenFold / OpenFold3 | Apache 2.0 | ✅ Yes |
| Chai-1 / Chai-2 | Apache 2.0 | ✅ Yes |
| Boltz-1 / Boltz-2 | MIT | ✅ Yes |
| Protenix | Apache 2.0 | ✅ Yes |
| AlphaFold 2 | Apache 2.0 | ✅ Yes |
| RF-AA | BSD-3 | ✅ Yes |
| RFdiffusion / RFdiffusion3 | BSD | ✅ Yes |
| AlphaFold 3 | Non-commercial (code public Nov 2024) | ❌ No |
6. The Path Forward
The protein AI ecosystem is moving fast. In the 15 months since AlphaFold's Nobel Prize, we've gone from "AF3 is locked behind a non-commercial license" to having multiple open-source alternatives that outperform it. The field is shifting from prediction to generation, designing new proteins, antibodies, and enzymes rather than just modeling known ones.
The biggest unsolved problems are downstream. We can predict structure well. We can design new proteins. But we're still bad at predicting function from structure, modeling the dynamics of how proteins move and interact in real cellular environments, and designing proteins that actually work when you synthesize them (wet-lab success rates are improving but still far from reliable). The gap between "model says this works" and "it actually works in a cell" is where most of the hard problems live.
For ML engineers, the most interesting near-term opportunity is probably at the intersection of generative models and experimental feedback. Models like ESM-3 and Chai-2 are starting to generate genuinely novel proteins, but closing the loop with experimental validation (active learning for protein design) is still early. The teams that figure out tight iteration between computation and wet-lab testing are going to have a massive advantage.
What's Next: Part II
In Part II (coming soon), I go from theory to practice: picking tools from this landscape, building an end-to-end protein AI pipeline, and training custom models. Code, benchmarks, and the failures along the way.
The future of medicine is code. I'm writing it in the open.
7. Key References
Foundational Papers
AlphaFold 2: Jumper, J. et al. "Highly accurate protein structure prediction with AlphaFold." Nature 596, 583–589 (2021). DOI
ESMFold: Lin, Z. et al. "Evolutionary-scale prediction of atomic-level protein structure with a language model." Science 379, 1123-1130 (2023). DOI
ProteinMPNN: Dauparas, J. et al. "Robust deep learning–based protein sequence design using ProteinMPNN." Science 378, 49-56 (2022). DOI
OpenFold: Ahdritz, G. et al. "OpenFold: Retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization." Nature Methods (2024). DOI
RFdiffusion: Watson, J.L. et al. "De novo design of protein structure and function with RFdiffusion." Nature 620, 1089–1100 (2023). DOI
ColabFold: Mirdita, M. et al. "ColabFold: making protein folding accessible to all." Nature Methods 19, 679–682 (2022). DOI
ESM-3: Hayes, T. et al. "Simulating 500 million years of evolution with a language model." Science (2025). DOI
Boltz-2: Wohlwend, J. et al. "Boltz-2: Exploring the Frontiers of Biomolecular Prediction." (2025). GitHub
RFdiffusion3: Watson, J.L. et al. "RFdiffusion3: De novo protein design with atom-level precision." (2025). GitHub
Protenix: ByteDance Research. "Protenix: An AI framework for protein structure prediction and beyond." (2026). GitHub
Architecture Deep-Dives
SE(3) Diffusion: Yim, J. et al. "SE(3) diffusion model with application to protein backbone generation." ICML (2023). arXiv
Folding Diffusion: Wu, K.E. et al. "Protein structure generation via folding diffusion." NeurIPS (2022). arXiv
LM-Design: Zheng, Z. et al. "Structure-informed Language Models Are Protein Designers." ICML (2023). arXiv
Part I of the OpenMed AI Biotech Series | February 2026
Part II coming soon: Building Your Own Protein AI Pipeline