
Consumer AI is scaling at historic speed without a comprehensive federal AI statute, even as the last two years brought governance blowups at leading labs, co-founder churn at xAI and Thinking Machines Lab, ongoing copyright litigation, and safety incidents involving consumer chatbots. The Trump Administration rescinded the Biden AI Executive Order within hours of taking office, and the 118th Congress introduced 150+ AI bills without producing a single binding, economy-wide standard. Washington and Wall Street are still writing checks, but the legal overhang is now explicit, with 72% of S&P 500 companies disclosing AI as a material risk.
AI for pharma and biotech is a different story. The FDA has issued guidance at roughly 3x its 2019-2023 annual rate, with a wave of major AI-related guidance landing in 2024-2025 alone. In parallel, the agency has deployed agentic AI internally and published joint principles with the EMA.
Regulation is not a tax on innovation; it is the underwriting layer. The FDA reduces deployment risk by standardizing what must be proven (endpoints, validation, monitoring) and how systems can change over time (controls, documentation, accountability). When the risk is bounded and repeatable, it becomes financeable.
What the data shows:
FDA medical product guidance output grew 5.8x in 35 years, with 82% of AI-specific guidance concentrated in 2024-2025
AI/ML devices went from 0.07% to ~8% of 510(k) clearances; the two largest accelerations followed framework-level regulatory events, not advances in AI capability alone
Drug development AI is entering the same regulatory build phase that devices passed through starting in 2017, but with a head start: frameworks like PCCP and GMLP established for devices may transfer directly, compressing the cycle
It is literally life or death. Pharma products go into human bodies, and failure is irreversible harm. You cannot ship a hotfix to a patient. That single fact forces higher proof standards, tighter controls, and less tolerance for iteration in production.

At these stakes, regulatory uncertainty is existential. A company cannot invest $500M in a Phase III trial wondering whether the FDA will accept its AI-generated endpoints. Deal terms reflect that: average upfront payments for Phase II leads jumped 460% from 2022 to 2024. AI discovery collaborations hit $29.7B in total deal value in 2025, with $800M in upfront payments, a 4x increase from the prior year. When that much capital is at stake, investors want rules, not a vacuum.
The lobbying data makes the contrast clean. The eight largest tech and AI companies spent a combined $36M on federal lobbying in H1 2025, about $320,000 per day Congress was in session. OpenAI alone increased lobbying 7x to $1.76M.
Consumer AI already operates under a permissive federal setup, so the priority is federal preemption to block state-level friction. Life sciences pushes in the other direction: stakeholders press the FDA to write clearer rules to de-risk capital, and the CDER AI Council now works with industry to publish Guiding Principles of Good AI Practice (GXP). Both sectors want federal action, but tech lobbies to prevent regulation and life sciences lobbies to create it.
The unit economics help explain the politics. Consumer AI monetizes through mass adoption and high-volume, low-dollar transactions, so onboarding friction and UX drag kill conversion. Pharma AI monetizes through a small number of institutional buyers underwriting nine-figure commitments, so regulatory friction functions as diligence and downside protection.
A caveat: “thriving” means the regulatory infrastructure works, not that every company wins. Recursion burned $370M in H1 2025 and merged with Exscientia in what the market read as consolidation, not strength, despite a $12B Roche partnership. Regulation built the playing field. Biology and unit economics still determine who survives on it.
The FDA broke down center silos and accelerated guidance output. This is not a bureaucracy expanding. It is an institution re-architecting itself to handle AI as a cross-cutting capability.
The FDA is not only regulating AI; it is piloting it internally. “Elsa,” an LLM-based system launched in May 2025, reached over 70% voluntary staff adoption. In December 2025, the FDA announced an agentic AI deployment for all agency employees, covering use cases including pre-market reviews, review validation, and post-market surveillance.
The deployment runs in a high-security GovCloud environment; models do not train on input data or industry submissions. Sponsors may reasonably expect submissions to face both human and machine-assisted scrutiny. That raises the value of structured, machine-readable submissions and internal pre-flight QA.
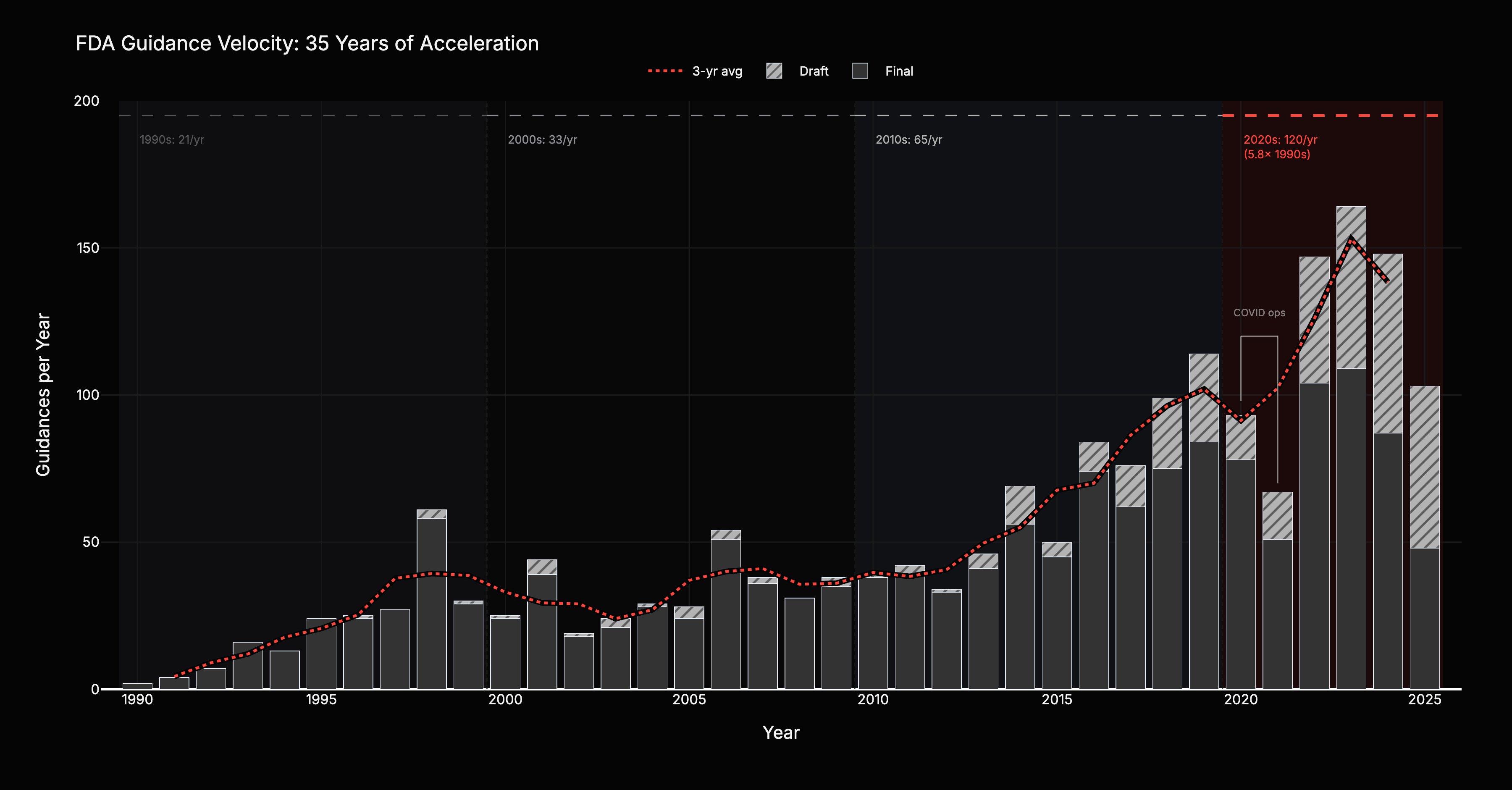
FDA guidance output, filtered to substantive medical product guidance, accelerated every decade for 35 years: from 21 per year in the 1990s to 33 in the 2000s (+57%), 65 in the 2010s (+97%), and 120 in the 2020s (+85%). A 5.8x increase. The unfiltered count, including food safety, tobacco, and compliance manuals, shows only 3.8x acceleration. Medical product guidance growth outpaced everything else. This is deliberate investment, not bureaucratic bloat.
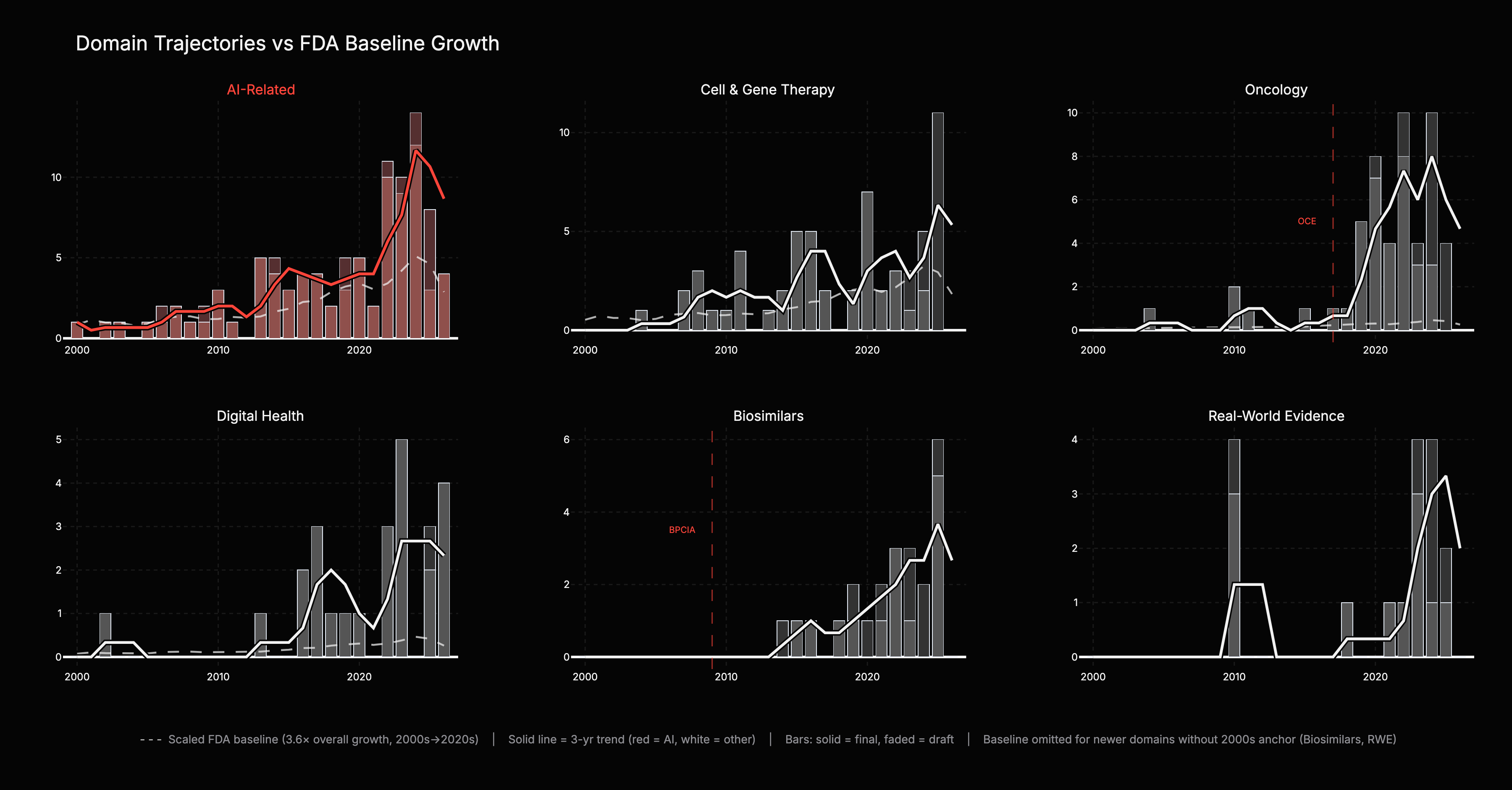
The six-panel chart answers the critical question: which domains genuinely outpace the FDA’s overall output growth, and which are just riding the tide? Each panel overlays a scaled baseline (dashed grey) showing what the domain’s trend would look like if it merely tracked the 3.6x overall medical product growth rate. The gap between the colored trend and the baseline is the signal.
AI-related guidance runs roughly 2x the baseline prediction, a strong signal driven by accumulation of many guidances rather than a single structural event. Digital Health had limited 2000s activity but its rapid acceleration tracks 2010s policy shifts. Real-World Evidence is an entirely new domain with no meaningful 2000s baseline; its growth a product of 2010s framework-building.
Oncology is the extreme case: 67x growth versus the 3.6x baseline, 19x above expected. Near-zero output in the 2000s, then rapid growth after FDA created the Oncology Center of Excellence in 2017. Biosimilars is the cleanest example: the domain did not exist before the Biologics Price Competition and Innovation Act in 2009, so every guidance in this panel traces back to statute. Inference: both illustrate what appears to be a repeating mechanism: new regulatory architecture precedes new markets.
For Biosimilars and Real-World Evidence, a pre-2010 baseline is not computable because the domains did not exist in the 2000s. Interpretation: rising regulatory output correlates with market vitality, not bureaucratic bloat. More developers building products creates more edge cases, and FDA responds with more rules. One plausible interpretation: guidance volume growth correlates with market vitality, up to a point: overregulation can freeze a market as effectively as underregulation. The current trajectory shows no signs of that constraint.
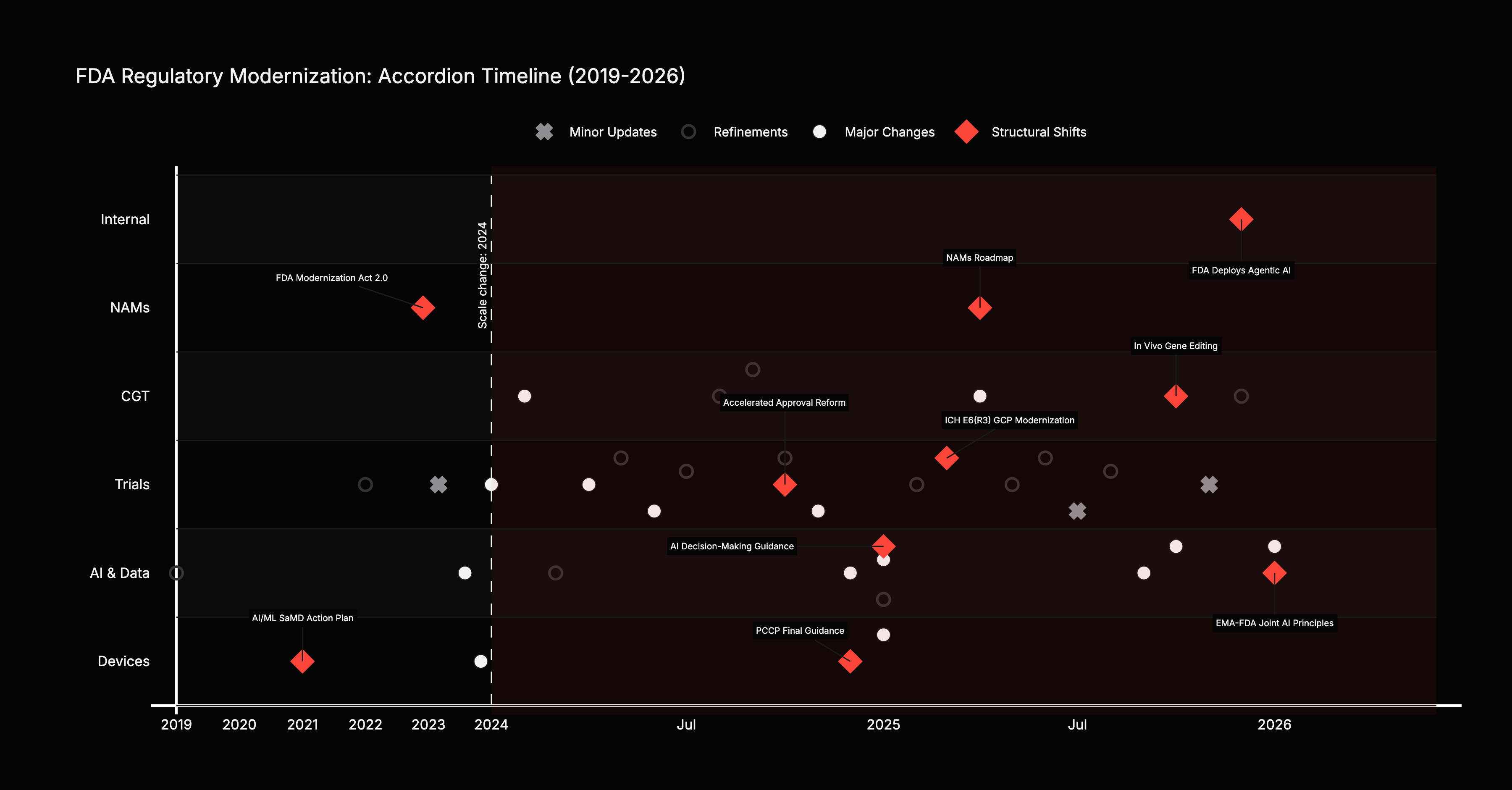
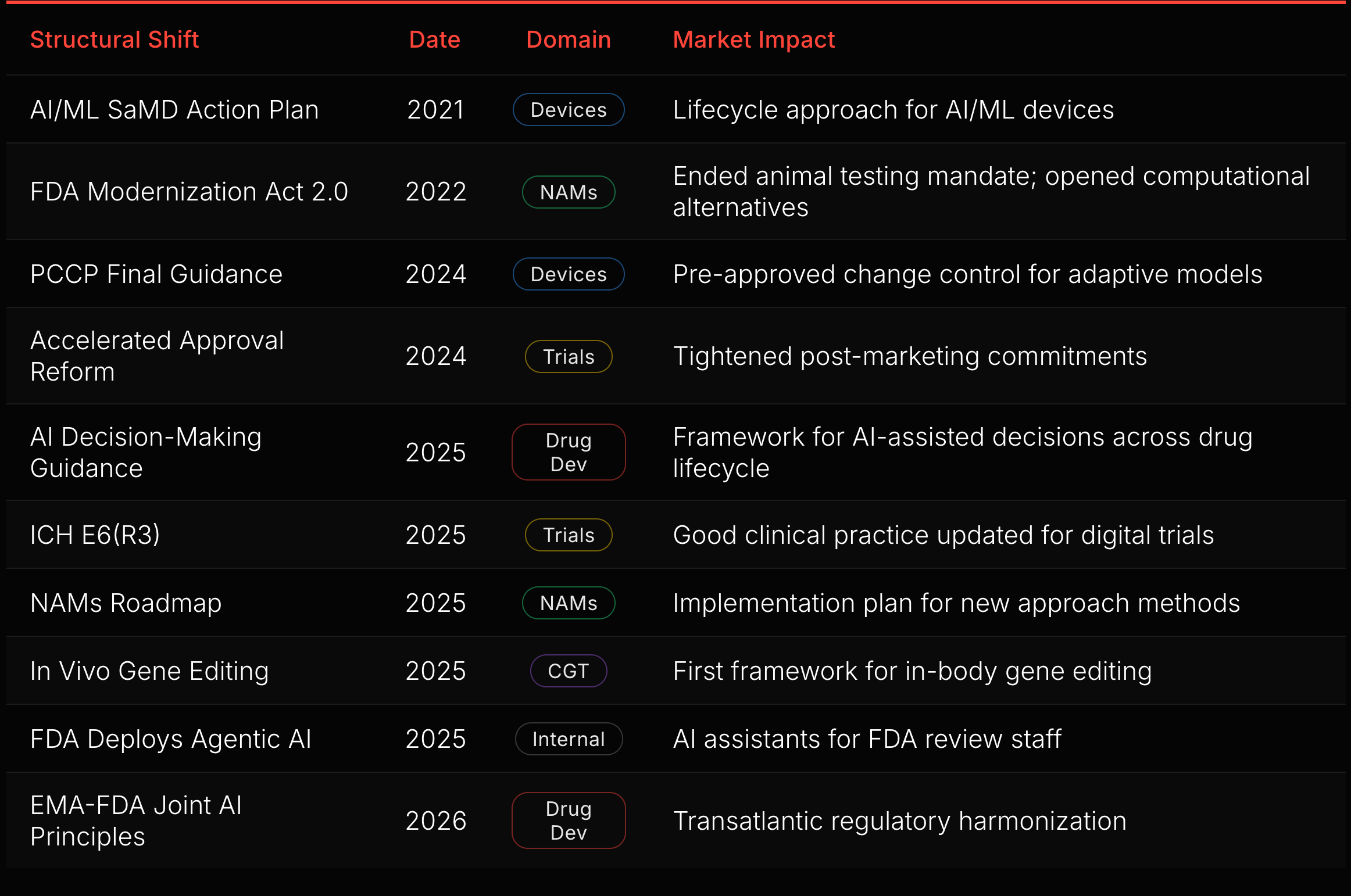
Forty-one curated regulatory actions mapped across five swimlanes. The density is the point: 82% came from 2024-2025. The framework for drug development AI is being laid right now, tracking the same pattern that devices followed starting in 2017.
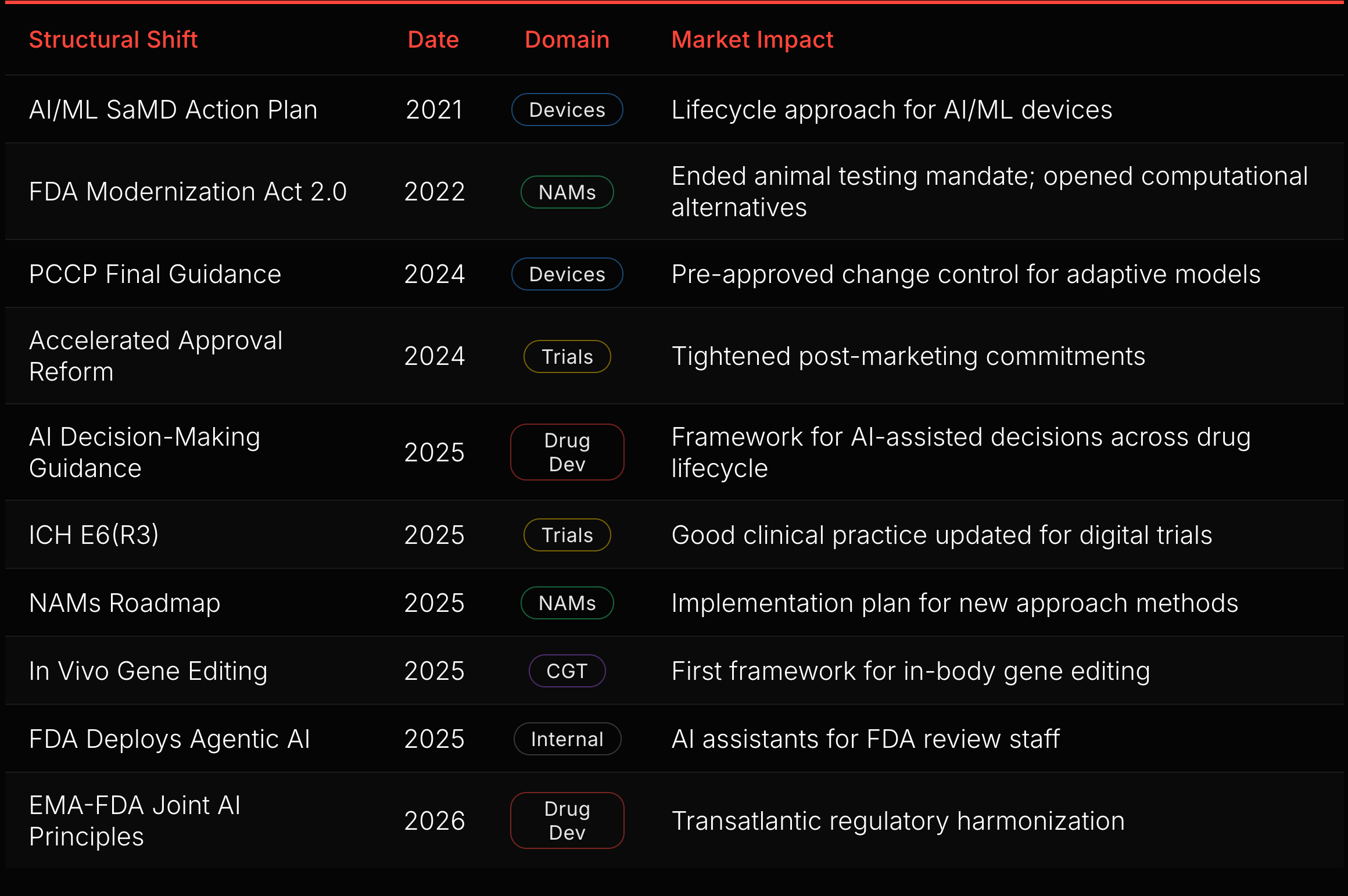
Ten of those 41 actions are structural shifts (red diamonds in Figure 5). Each changes the rules for an entire domain. The sequence reveals the FDA’s construction order.
The device foundation came first. The SaMD Action Plan (2021), GMLP (2021), and PCCP Final Guidance (2024) built a complete regulatory stack for AI/ML devices: lifecycle approach, development principles, and pre-approved change control. That stack is now the template for drug development (see “AI Medical Devices” below).
The drug development build-out started in 2025. The AI Decision-Making Guidance addresses how FDA will evaluate AI-assisted decisions across the drug lifecycle, from target identification through post-market surveillance. The EMA-FDA Joint AI Principles, published in January 2026, will extend this framework transatlantically.
Trial infrastructure is modernizing in parallel. Accelerated Approval Reform (2024) tightened post-marketing commitment enforcement, closing a gap where sponsors delayed confirmatory trials. ICH E6(R3) (2025) updated Good Clinical Practice for digital trials, electronic consent, and decentralized endpoints. These reshape the infrastructure that AI tools operate within.
New modalities created new regulatory surface. The FDA Modernization Act 2.0 (2022) ended the mandatory animal testing requirement. The NAMs Roadmap (2025) followed with an implementation plan. In vivo gene editing guidance (2025) established the first framework for a domain that did not exist a decade ago.
The FDA itself is transforming. Deploying agentic AI for its own review staff changes review timelines, consistency, and the agency’s capacity to process rising submission volume.
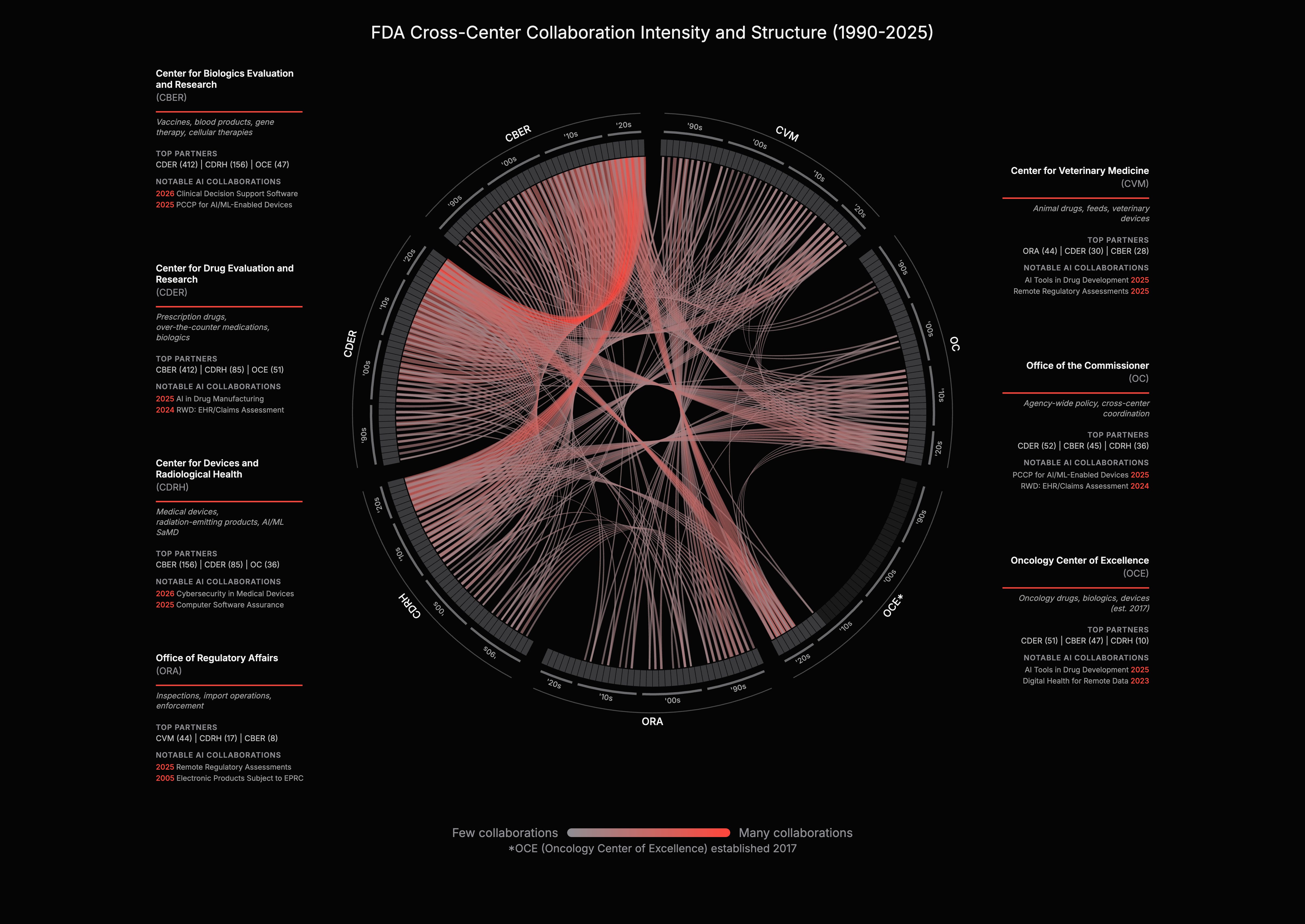
FDA centers historically operated independently. CDER handled drugs, CDRH handled devices, CBER handled biologics. AI cuts across all of them. Today, 27% of all guidances involve two or more FDA centers. When the FDA published "How CBER, CDER, CDRH, and OCP Work Together" on AI in 2024, it formalized what the data already showed: the agency is reorganizing around AI.
For companies, this means lower regulatory variance. Cross-center standardization could also enable cross-domain data flow: a CDRH-cleared device generating continuous patient data could, in principle, inform CDER drug development endpoints and monitor response during a trial. A company using AI across drug development and companion diagnostics would work under one framework, cutting the cost and timeline of multi-product submissions and allowing it to port medical device precedent into drug applications. The PCCP guidance that enabled pre-approved AI model updates for devices is a template, not a one-off.
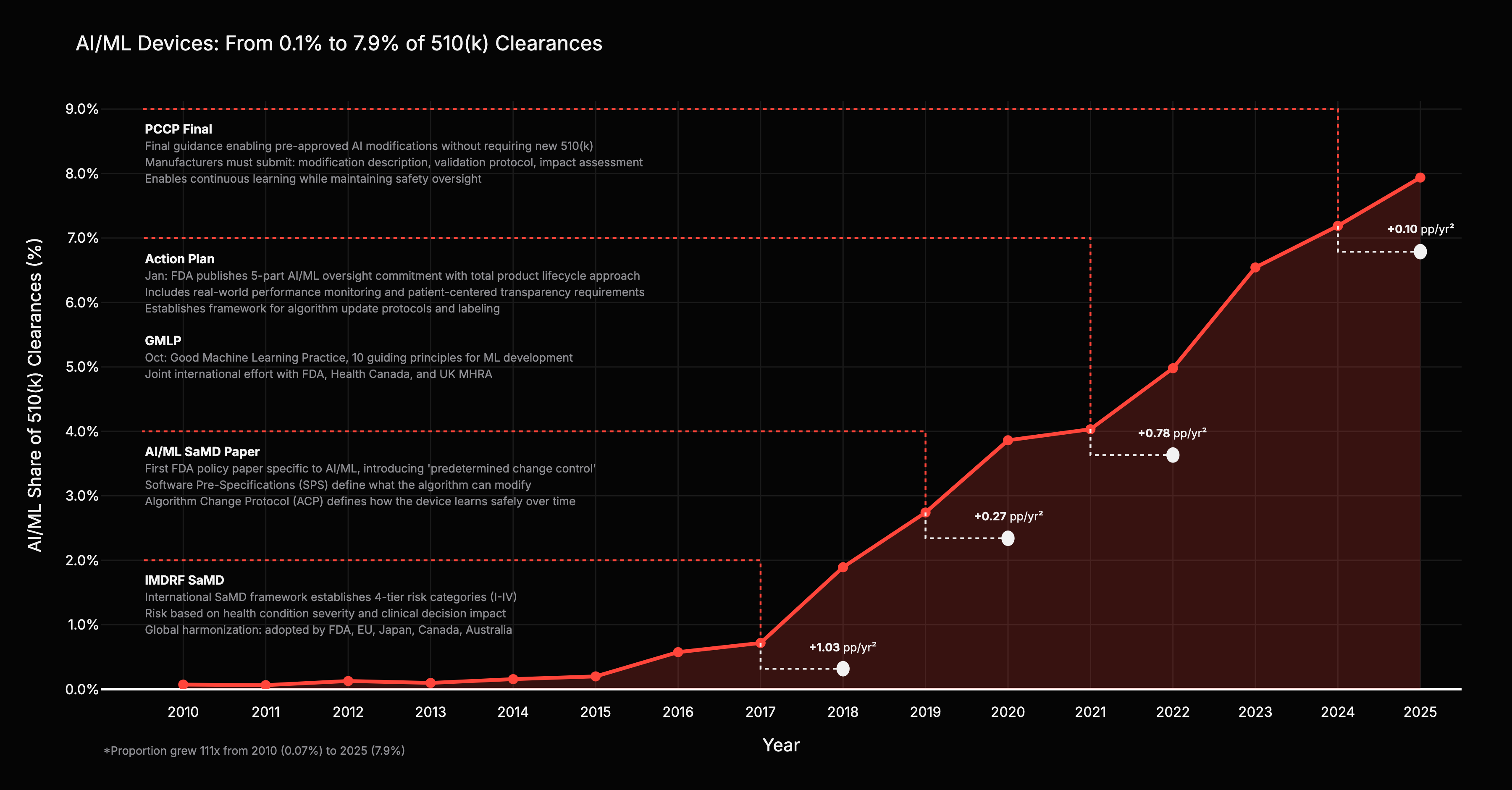
AI/ML’s share of total 510(k) clearances grew from 0.07% in 2010 to nearly 8% in 2025, a 111x increase in regulatory engagement share. The total device market grew; AI/ML grew faster.
The acceleration coincides with specific regulatory events, not just the march of AI capability. After the IMDRF SaMD framework in 2017, AI/ML’s share accelerated by +1.03 percentage points per year squared. After the Action Plan and GMLP in 2021, another +0.78 pp/yr^2. The two largest jumps followed the two framework-level guidances. Discussion papers and narrow technical updates barely moved the needle.
Inference: the consistent +1 year lag between guidance publication and application surge points to a regulatory-capital channel: regulators publish, companies adjust investment, applications follow. Regulation was necessary, not sufficient. The transformer architecture (2017) and GPU democratization provided the engine. But without a pathway to clear stochastic algorithms, capital would have frozen due to liability risk.
1,357 AI/ML medical devices have been authorized through 2025. Authorizations show step changes tied to regulatory releases: a clear jump in 2017-2018 after IMDRF SaMD, continued growth in 2019-2020, a COVID-era plateau in 2020-2021, renewed growth in 2021-2023 after the SaMD Action Plan and GMLP, then a 2024 pause with an uptick after the PCCP Final.
The IMDRF SaMD framework in 2017 was the first inflection. It established four-tier risk categories (I-IV) based on the severity of the health condition and the weight of the clinical decision, adopted globally by the FDA, EU, Japan, Canada, and Australia. For the first time, companies knew how to classify AI/ML software products. Investment followed: digital health funding grew from roughly $4.5B in 2015 to over $29B by 2021.
The SaMD Action Plan and GMLP in 2021 gave developers confidence that adaptive AI models would have a regulatory pathway, introducing a total product lifecycle approach and 10 guiding principles for ML development. The PCCP final guidance in 2024 clarified a pathway for pre-specified model changes without a new submission, subject to an agreed plan, validation, and reporting.
Radiology dominates at 77% of authorizations. Two factors likely contribute: radiology is imaging-native and non-invasive, so training data proliferates at scale; and imaging endpoints are well-defined, allowing AI to integrate into existing workflows with minimal disruption. Interpretation: data availability and clear validation endpoints appear to be preconditions for AI device commercialization, not just regulatory clearance. The “Other” category growing visible in 2023-2025 suggests specialty diversification is finally happening.
One important caveat: authorization does not equal adoption. Pear Therapeutics raised over $400M, secured FDA clearance for digital therapeutics treating substance abuse, and filed for bankruptcy in 2023 because it had regulatory clearance but no reimbursement pathway. The lesson segments by category. Diagnostics and digital therapeutics are tightly coupled to reimbursement and care pathways; clearance alone is insufficient. Upstream drug discovery AI is less reimbursement-constrained but still bound by evidentiary standards downstream.
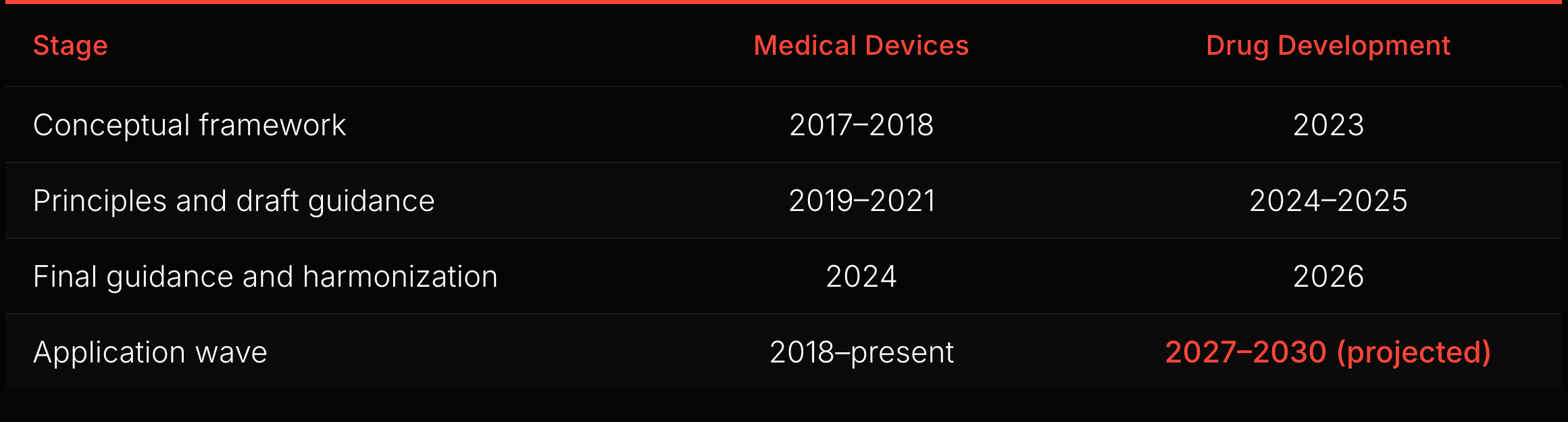
The analogy has limits. Devices commercialize through a shorter regulatory-to-market cycle: authorization, reimbursement coding, and procurement. Drugs face a longer chain: approval, reimbursement negotiation, formulary inclusion, clinical adoption, and label lifecycle management. The downstream complexity differs in degree, not in kind. But the upstream mechanism transfers: framework-level guidance converts regulatory uncertainty into calculable risk, unlocking capital deployment.
If the observed device lag is a useful analog, the next measurable inflection in drug-development AI submissions could appear within the next couple of years. The starting gun may already have fired: Insilico Medicine’s rentosertib, the first generative AI-designed drug to complete a Phase IIa trial, posted positive efficacy results in Nature Medicine in 2025. The indicators to watch: draft-to-final cadence on AI drug development guidances, the number of IND submissions referencing AI/ML methodologies, growth in ISTAND pilot acceptances, and uptake of Bayesian methodology under the January 2026 guidance. The 2024-2026 guidance wave is building the regulatory infrastructure. Whether the application wave follows at the same lag depends on these signals.

The FDA leads across both dimensions and is among the few agencies publicly describing agency-wide internal AI deployment. The EMA (EU) has the strongest real-world evidence infrastructure (DARWIN EU: 180M active patients across 16 countries). The MHRA (UK) runs the most aggressive sandbox approach globally (AI Airlock with 5 devices in pilot). The MFDS (South Korea) is among the first countries with LLM-specific regulatory guidance.
The EU presents a unique challenge. The EU AI Act adds a “horizontal” compliance layer on top of existing “vertical” health regulations, a complexity absent in the FDA’s centralized approach. Medical device AI in Europe now faces a dual burden: products must satisfy both the clinical safety requirements of the MDR and the high-risk classification requirements of the AI Act, creating distinct but overlapping compliance timelines.
China’s NMPA is pursuing a “standardization-first” strategy. Rather than sandbox models, the NMPA aggressively defines classification catalogs for AI software and enforces standard datasets for registration, aiming to streamline local market entry through rigid technical benchmarks. For jurisdictions without sovereign AI guidance, the WHO has stepped in as the global coordinator, releasing foundational guidance on Large Multi-modal Models in January 2024 that sets a de facto governance baseline for low- and middle-income countries.
Device AI regulation is more mature than drug AI across every jurisdiction. The 5-year gap observed at the FDA exists globally. International convergence is accelerating through EMA-FDA Joint Principles, IMDRF GMLP, and ICH M15, but remains incomplete. Ten-plus jurisdictions still release overlapping, non-identical guidance.
The implication: companies with FDA-first regulatory strategies have the clearest path. International harmonization means FDA alignment travels globally. Companies engineering multi-jurisdiction submissions can shorten timelines by reducing duplicated regulatory work.
Five hypotheses the regulatory data supports.
1. Deploy during the draft-to-final window. Capital follows regulatory clarity, not just scientific breakthrough. The NAMs Roadmap preceded a $2.7B organoids market. DCT guidance preceded $500M+ in platform VC. Biosimilar streamlining preceded $232B in addressable biologics opening up, 90% without competition. Correlation is not causation, but the pattern is consistent: guidance drops, regulatory uncertainty declines, capital deployment accelerates. The current windows are biosimilars (2024-2025 guidance), NAMs (April 2025), and Bayesian methodology (January 2026). The Bayesian guidance allows sponsors to “borrow” prior information for primary inference, which has the potential to reduce Phase II/III sample sizes, a direct cap-ex reduction event.
2. Regulation is a capital signal, not a barrier. The device data: 111x regulatory engagement growth, with the two largest accelerations following framework-level guidance. The drug development AI market is entering the same phase. The FDA’s ISTAND pilot program validates non-traditional drug development tools that do not fit standard regulatory buckets. Acceptance into ISTAND functions as a platform-level analog to Fast Track designation: it signals regulatory engagement before a single drug candidate enters the clinic. The analogy is in investor signaling value, not statutory benefit.
3. The “Two FDAs” tension comes into focus. The FDA is absorbing political skepticism around traditional biologics, including vaccines, while accelerating AI framework-building. This is not a clean contradiction, but “prioritization” understates the tension. It is governance under cross-pressures.
Model the FDA as two coupled systems: (1) modality politics, high variance, driven by administration priorities and public sentiment; (2) platform infrastructure, lower variance, driven by institutional continuity and technical necessity. AI-tool validation largely sits in system (2).
The evidence supports this framing. AI regulatory framework-building now has bipartisan institutional momentum spanning the Biden and Trump administrations. Despite a 10-for-1 deregulatory executive order, nearly 90% senior leadership turnover, and net losses of 1,093 employees at CDER and 224 at CBER in FY 2025, the FDA continued issuing AI guidance documents. That continuity lowers tail risk for AI tool validation and review pathways. Phase II deal premiums are up 460%. AI discovery upfronts quadrupled year-over-year. Capital is pricing in a clearer route to regulatory acceptability for AI-enabled development.
At the same time, the current administration has narrowed guidance in specific therapeutic areas, most visibly vaccines, even as AI frameworks advance. Inference: the AI track benefits from institutional momentum independent of political direction, but the broader FDA operates under unusually high political pressure in other domains. The staffing losses are a leading indicator: if review timelines lengthen in FY 2026 user fee reports, the capacity constraint becomes material. Investors should model this as complexity with asymmetric impacts by modality, not a resolved paradox.
4. The “Digital Biomarker” alpha. The FDA’s DHT framework has opened a pathway for endpoint engineering. Capital should flow not just to faster trials, but to trials made possible by newly measurable endpoints. Companies validating novel digital endpoints, gait analysis for Parkinson’s, voice biomarkers for Alzheimer’s, are creating new addressable markets for drugs that previously failed due to insensitive measurement tools. The measurement moat is becoming as valuable as the molecule.
5. The ISTAND signal. Investors watch Fast Track designation as a molecular de-risking event. For AI platforms, ISTAND acceptance may serve as an analogous signal: regulatory engagement that de-risks the platform technology before a single candidate enters the clinic. This is the leading indicator for platform value.
The “Glass Box” premium. The FDA’s 2023 discussion paper on AI/ML emphasizes that explainability is a safety requirement, not a feature. This suggests a future valuation gap: “Glass Box” models with built-in interpretability and mechanistic grounding will command a premium over “Black Box” competitors. The market will discount high-performance models that cannot explain their predictions in a 21 CFR Part 11 compliant audit trail.
The “Infrastructure as Regulation” trap. Is the regulatory AI market a standalone category or a feature absorbed by Veeva and IQVIA? Most startups are building speed moats. Speed moats decay. Data moats endure. Interoperability moats are defensive. The winner may not be the best AI model, but the one that integrates seamlessly with the FDA’s new digital audit infrastructure.
The “Data Sovereignty” border. The EU AI Act classifies medical AI as High Risk, with penalties up to 7% of global revenue. US models trained on non-representative data may perform well technically but fail legally in Europe. This is no longer a compliance issue; it is a data sovereignty question. A speed moat in the US is worthless in the EU if the model is legally inadmissible in the EMA’s jurisdiction.
Regulation builds defensible assets. Deregulation builds cash flow velocity. Consumer AI is generating historic revenue at historic risk. That is a trade, not an investment. The FDA’s 2026 roadmap offers the inverse: high-friction validation that creates high-value, defensible interoperability. Part 2 of this article explores where the capital should flow.
[1] White House, “Removing Barriers to American Leadership in AI” (EO 14179, Jan 2025). https://www.whitehouse.gov/presidential-actions/2025/01/removing-barriers-to-american-leadership-in-artificial-intelligence/
[2] Brennan Center for Justice, “Artificial Intelligence Legislation Tracker” (118th Congress). https://www.brennancenter.org/our-work/research-reports/artificial-intelligence-legislation-tracker
[3] PBS, “Senate Pulls AI Regulatory Ban from Big Beautiful Bill.” https://www.pbs.org/newshour/politics/senate-pulls-ai-regulatory-ban-from-gop-bill-after-complaints-from-states
[4] Mintz, “Trump Signs Law with Over $1B in AI Funding.” https://www.mintz.com/insights-center/viewpoints/54731/2025-07-11-president-trump-signs-law-over-1-billion-ai-funding-and
[5] FDA, “FDA Expands AI Capabilities: Agentic AI Deployment” (Dec 2025). https://www.fda.gov/news-events/press-announcements/fda-expands-artificial-intelligence-capabilities-agentic-ai-deployment
[6] EMA/FDA, “Guiding Principles of Good AI Practice in Drug Development” (Jan 2026). https://www.ema.europa.eu/en/news/ema-fda-set-common-principles-ai-medicine-development-0
[7] FDA, “Artificial Intelligence-Enabled Medical Devices.” https://www.fda.gov/medical-devices/software-medical-device-samd/artificial-intelligence-enabled-medical-devices
[8] FDA, “PCCP Marketing Submission Guidance” (Dec 2024). https://www.fda.gov/regulatory-information/search-fda-guidance-documents/marketing-submission-recommendations-predetermined-change-control-plan-artificial-intelligence
[9] Deloitte, “Measuring Return from Pharmaceutical Innovation” (15th Annual). https://www.deloitte.com/us/en/Industries/life-sciences-health-care/articles/measuring-return-from-pharmaceutical-innovation.html
[10] ASPE/HHS, “Drug Development.” https://aspe.hhs.gov/reports/drug-development
[11] BIO, “Clinical Development Success Rates 2011-2020.” https://www.bio.org/clinical-development-success-rates-and-contributing-factors-2011-2020
[12] BioSpace, “Top Biopharma Licensing Deals 2024.” https://www.biospace.com/business/the-top-7-biopharma-licensing-deals-of-2024
[13] Nature Biotechnology, “Deal Trends 2025.” https://www.nature.com/articles/d43747-025-00113-2
[14] Issue One, “Big Tech Lobbying.” https://issueone.org/articles/as-washington-debates-major-tech-and-ai-policy-changes-big-techs-lobbying-is-relentless/
[15] MIT Technology Review, “OpenAI Ups Its Lobbying Nearly Seven-Fold.” https://www.technologyreview.com/2025/01/21/1110260/openai-ups-its-lobbying-efforts-nearly-seven-fold/
[16] FDA Guidance Documents database (Excel export, 2,560 records after cleaning). See scripts/README.md for methodology.
[17] FDA Oncology Center of Excellence, established 2017.
[18] Biologics Price Competition and Innovation Act (BPCIA), enacted 2009 as part of the Affordable Care Act.
[19] Curated dataset of 41 regulatory actions from FDA guidance documents, 2019-2026.
[20] FDA, “How CBER, CDER, CDRH, and OCP Work Together on AI” (2024). https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-use-artificial-intelligence-support-regulatory-decision-making-drug-and-biological
[21] EMA, “DARWIN EU Expansion.” https://www.ema.europa.eu/en/news/darwin-eur-continues-expanding-its-capacity-deliver-real-world-data-studies
[22] MHRA, “AI Airlock Regulatory Sandbox” (May 2024). https://healthcarelifesciences.bakermckenzie.com/2024/05/10/mhra-launches-ai-airlock-to-address-challenges-for-regulating-medical-devices-that-use-artificial-intelligence/
[23] Korean MFDS GenAI Guidelines. https://pmc.ncbi.nlm.nih.gov/articles/PMC12123075/
[24] IMDRF, “Good Machine Learning Practice Finalized” (2025). https://www.raps.org/news-and-articles/news-articles/2025/1/imdrf-finalizes-good-machine-learning-practice,-so
[25] ICH M15, “General Principles for Model-Informed Drug Development.” https://www.ema.europa.eu/en/ich-m15-guideline-general-principles-model-informed-drug-development-step-2b-scientific-guideline
[26] UK-Singapore Regulatory Innovation Corridor. https://www.gov.uk/government/news/uk-and-singapore-launch-a-regulatory-innovation-corridor-to-speed-up-access-to-breakthrough-health-technologies
[27] Grand View Research, “Human Organoids Market Report.” https://www.grandviewresearch.com/industry-analysis/human-organoids-market-report
[28] Medable Series D Funding. https://hitconsultant.net/2021/10/26/medable-dct-series-d-funding/
[29] Center for Biosimilars, “New FDA Guidance Removing Barriers.” https://www.centerforbiosimilars.com/view/a-closer-look-at-new-fda-guidance-removing-barriers-to-biosimilar-development
[30] Medidata SCA (36K trials, 11M patients): https://www.3ds.com/newsroom/press-releases/medidata-trinetx-and-datavant-partner-enable-seamless-integration-real-world-data-clinical-development
[31] Cranium, “EU AI Act August 2025 GPAI Compliance.” https://cranium.ai/resources/blog/navigating-the-eu-ai-act-august-2025-deadline-gpai-compliance-penalties-and-enforcement/
[32] Fortune, “Cursor has $1B+ in revenue and a $29B valuation.” https://fortune.com/2025/12/11/cursor-ipo-1-billion-revenue-brainstorm-ai/
[33] Business Insider, “Midjourney revenue and team size.” https://www.businessinsider.com/midjourney-500-million-revenue-40-employees-generative-ai-2024-12
[34] BioPharma Dive, “Recursion-Exscientia merger.” https://www.biopharmadive.com/news/recursion-exscientia-merger-ai-drug-discovery/724618/
[35] openFDA, “Device 510(k) API.” https://open.fda.gov/apis/device/510k/
[36] Reed Smith, “The EU AI Act and Medical Devices: Navigating High-Risk Compliance,” 2025.
[37] Cisema, “China’s 2025 Medical Device Industry Standard Formulation and Revision Plan,” April 2025.
[38] WHO, “Ethics and governance of artificial intelligence for health: Guidance on large multi-modal models,” January 2024.
[39] FDA, “Framework for the Use of Digital Health Technologies in Drug and Biological Product Development,” Guidance for Industry, March 2023.
[40] FDA CDER, “Innovative Science and Technology Approaches for New Drugs (ISTAND) Pilot Program,” December 2023 Update.
[41] FDA, “Using Artificial Intelligence & Machine Learning in the Development of Drug and Biological Products,” Discussion Paper, May 2023.
[42] Code of Federal Regulations, Title 21, Part 11 (Electronic Records; Electronic Signatures).
[43] Rock Health, “2021 Year-End Digital Health Funding: Seismic Shifts Beneath the Surface,” 2022. https://rockhealth.com/insights/2021-year-end-digital-health-funding-seismic-shifts-beneath-the-surface/
[44] Pharmaceutical Technology, “Pear Therapeutics: A Lesson for Future DTx Developers,” 2023. https://www.pharmaceutical-technology.com/analyst-comment/pear-therapeutics-a-lesson-for-future-dtx-developers/
[45] PBS News, “Sam Altman reinstated as OpenAI CEO with new board replacing the one which fired him” (Nov 2023). https://www.pbs.org/newshour/nation/sam-altman-reinstated-as-openai-ceo-with-new-board-replacing-the-one-which-fired-him
[46] Fortune, “Half of xAI’s founding team has left, potentially complicating Elon Musk’s SpaceX IPO plans” (Feb 2026). https://fortune.com/2026/02/11/half-of-xai-founding-team-has-left-elon-musks-ai-company-potentially-complicating-his-plans-for-a-blockbuster-spacex-ipo/
[47] TechCrunch, “Mira Murati’s startup, Thinking Machines Lab, is losing two of its co-founders to OpenAI” (Jan 2026). https://techcrunch.com/2026/01/14/mira-muratis-startup-thinking-machines-lab-is-losing-two-of-its-co-founders-to-openai/
[48] NPR, “’The New York Times’ takes OpenAI to court. ChatGPT’s future could be on the line” (Jan 2025). https://www.npr.org/2025/01/14/nx-s1-5258952/new-york-times-openai-microsoft
[49] NPR, “Lawsuit: A chatbot hinted a kid should kill his parents over screen time limits” (Dec 2024). https://www.npr.org/2024/12/10/nx-s1-5222574/kids-character-ai-lawsuit
[50] Harvard Law School Forum on Corporate Governance, “AI Risk Disclosures in the S&P 500: Reputation, Cybersecurity, and Regulation” (Oct 2025). https://corpgov.law.harvard.edu/2025/10/15/ai-risk-disclosures-in-the-sp-500-reputation-cybersecurity-and-regulation/
[51] Insilico Medicine et al., “A generative AI-discovered TNIK inhibitor for idiopathic pulmonary fibrosis: a randomized phase 2a trial,” Nature Medicine (Jun 2025). https://www.nature.com/articles/s41591-025-03743-2
[52] European Parliament and Council, Regulation (EU) 2024/1689 (EU AI Act), Official Journal of the European Union (Jul 2024). https://eur-lex.europa.eu/eli/reg/2024/1689/oj/eng
[53] BioSpace, "Opinion: 2025 Reshaped the FDA. What Will 2026 Hold?" (Jan 2026). https://www.biospace.com/fda/opinion-2025-reshaped-the-fda-what-will-2026-hold
[54] Anthropic, "Anthropic raises $30 billion Series G funding, $380 billion post-money valuation" (2025). https://www.anthropic.com/news/anthropic-raises-30-billion-series-g-funding-380-billion-post-money-valuation