
One of our most recent updates at Carolina Cloud is adding Sentieon to our Genomics Container. Sentieon is a licensed software package that accelerates the standard genomics pipeline from FASTQ to VCF. The acceleration is achieved through mathematical optimization of algorithms and improved resource management i.e better parallelization, plus they have some clever machine learning models. To be honest I had not heard of it before speaking to Victory Genomics, an equine genomics company with whom we recently started working. Victory uses cutting edge genomics to provide insights into horse genetics across multiple disciplines. Unlike other equine genomics companies, they use whole genome sequences analyzed by top-level human medical geneticists and bioinformaticians, yielding far superior and detailed results. Naturally I was excited to try Sentieon out, a good excuse to do some genomics analysis on the job.
Because we were working with an equine genomics company I decided to do some benchmarking with horse genomes. There are plenty of publicly available high coverage samples, mostly out of Kentucky. For my high coverage sample I used a publicly available 21x genome from a Debao pony, a tiny little cute breed from china. Details on the reference sequence, sample accessions and data acquisition are available in the Appendix of this post.
For the compute setup I used Carolina Cloud Genomics Containers. These are simply great for benchmarking for a number of reasons:
All the necessary bioinformatics (and now Sentieon) tools come preinstalled meaning no installation time waste.
They always have dedicated CPUs so each run is guaranteed the same compute access. No CPU steal messing up our results.
Live container resize ability means we can try out different resource amounts systematically without having to create new instances and move data between them. When working with 100s of GiB of genomics data this is a huge time and efficiency saver. NOTE to be really safe you want to drop the cache, save and remove previous run output files and maybe even restart the container even though we can live resize to make sure each run is a cold start. But this pales into comparison to the hassle of making new instances with different resources.
The UI resource utilization viewer gives you a pretty look at the live resource use as your pipeline is running, far better than
sstatorseff(if you know you know).Seamless object storage integration with
aws clipreinstalled and bucket environment variables automatically preset on container spin-up means moving files to storage could not be easier from the get go.
With my sales pitch out the way, here are some important details about this project; All genomics containers have OS Linux Ubuntu 24.4 and provisioned with 1/2T of local disk storage. For each run we use the DNAscope tool that does alignment, sorting, removing/marking duplicates and variant calling wrapped in a bash script (see Appendix). We used the same input files and the same parameters except those we were testing and the runtime for each run was measured with the time command.
We are actually going to do a cinematic jump to the end of the story and give the final results table:
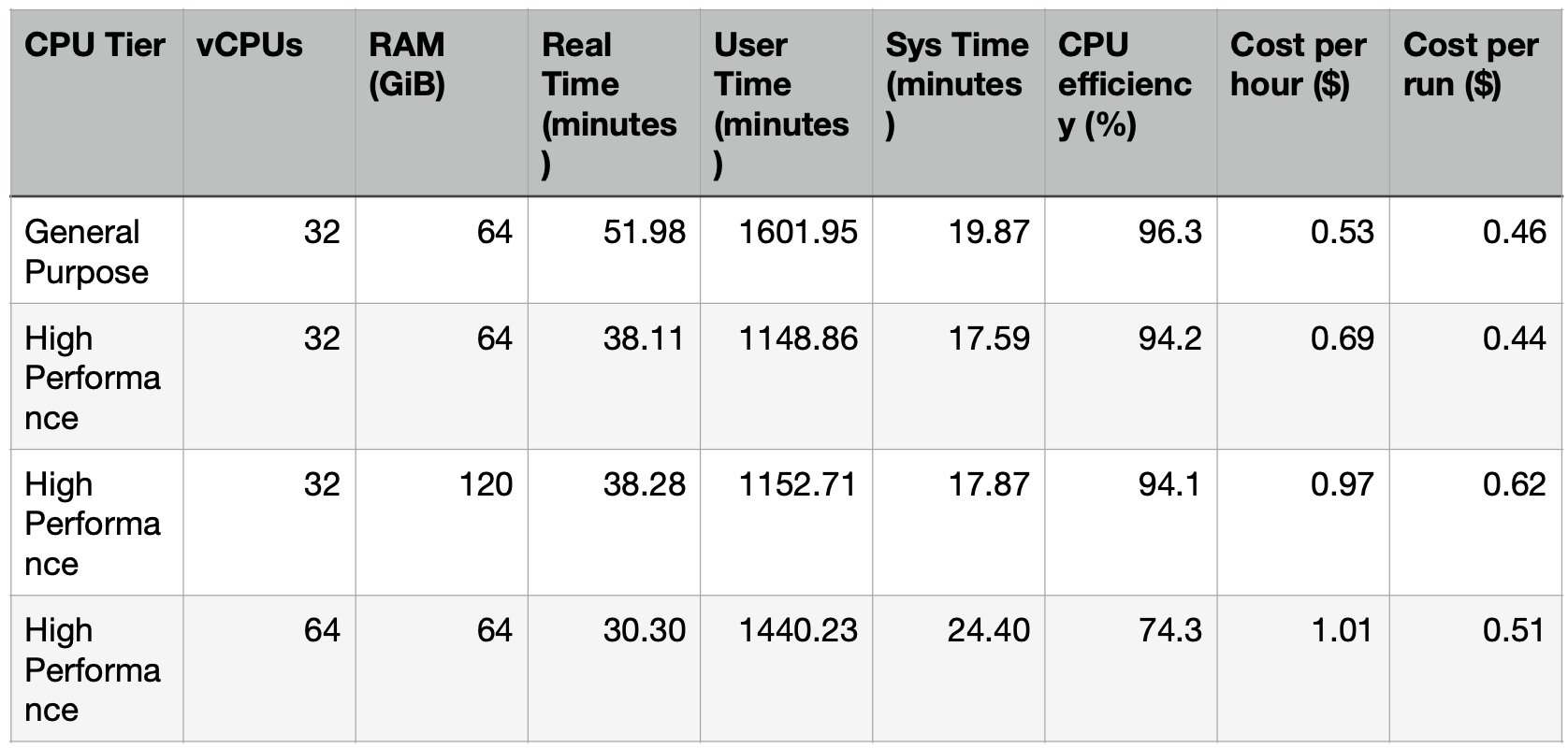
I want to point out and overall result now, a scene at the end of the movie that makes the whole story more credible; Sys time is CPU time spent inside the OS kernel serving your program whereas User time is the CPU time spent running your program’s own code. Essentially a small Sys time compared to User suggests that the pipeline is compute-bound rather that I/O-bound or bogged down with filesystem/network operations. Most of the CPU time is spent actually running Sentieon which is what we like.
Ok, cinematic lurch back to the beginning and lets make this ending make sense. I started on one of our General Purpose machines, an AMD EPYC 9755 QS with clock speed 3.1GHz boost. With a 21x genome I took Sentieon’s recommended 32 vCPU and 64 GiB RAM and hit play. From FASTQ to VCF it finished in just under 52 minutes (Real time) which is a significant speed up compared to using BWA-MEM and GATK alone. What is more, is that running this pipeline required no next level expertise where usually we would need to split by chromosome or smaller regions in order to parallelize effectively. All I had to do was change the `nt` bash variable and Sentieon did all that for me.
So next I want to see how much faster it would run on a High Performance machine, an AMD Threadripper 7975WX with clock speed 5.3GHz boost. It finished in just over 38 minutes and while the hourly price of the higher clock speed machine is bigger, this speedup meant that the cost per run came out to about the same as the General Purpose run. Going with High Performance is therefore a no brainer; get 37 genomes finished in one day vs 27 for the same if not slightly cheaper cost.
Sticking with the High Performance CPU I started to play with the resources via live container resize. First I doubled the RAM to 120GiB and saw no significant change in time only an increase in cost. For a 21x genome, not worth it.
What about vCPUs? I upped the number to 64 keeping RAM at 64GiB resulting in a decrease in runtime to just over 30 minutes. That’s about a 20% speed up in runtime per genome for a 16% increase in cost per run. Not a no brainer but not an outright no. If someone is going for speed over budget, your client wants his/her horse genomes done by the next time their thoroughbred races, then increasing vCPUs to get 47 genomes done per day might be a good option. If budget is more of a concern then maybe not.
But what is actually happening here? Why does doubling the vCPUs not half the runtime? Why is Sentieon not taking care of full parallelization. Those of you detail hunters will be looking now at the CPU efficiency column. Where before at 32 vCPUs we were at near enough full efficiency, now at 64 we see a drop to 74.3%. CPU efficiency is calculated as User time / Real time * Number of vCPUs, so when all the CPUs are being used for all the wall clock time this value is 1 or 100%. Less than 100% and some of the CPUs are sitting idle for some time.
Sitting with my coffee, I was mesmerized by the pretty resource utilization panel on my Carolina Cloud instance card and I noticed the change. CPU usage hovered around 70% for the alignment phase, while the variant calling stage had near 100% usage throughout. As these are the two biggest stages of the pipeline we average out at around 75% efficiency. (Note these are not pinned cores but all 64 vCPUs being used at 70% capacity). Alignment does not parallelize perfectly even with Sentieon, leading to diminishing returns as vCPU count increases and wasted energy per genome as well as money. This behavior is consistent with Amdahl’s Law which describes mathematically how speedup of a system is limited by the non-parallelizable fraction. Fittingly Amdahl’s first name was Gene!
That brings us back around to the end of the benchmarking story. There is obviously lots of further tests we could do, some of which I will mention below. Here are my key takeaways in no particular order:
Sentieon is great, delivering a substantial reduction in runtime compared to traditional BWA+GATK pipelines. Their algorithms are compute-bound, with low sys time and high CPU efficiency without engineering overhead of chunking chromosomes into regions and then concatenating.
Clock speed was the biggest win on Carolina Cloud instances, where a 26% decrease in runtime was achieved at no extra cost per run by switching to the High Performance CPU range. Going to 120GiB RAM produced no gain for our 21x genomes but at higher coverages 64GiB might be limiting. A nice extension would be so see how far down in RAM we could go (and therefore cost per run) before we see memory limitations.
More vCPUs yields diminishing returns consistent with Amdahl’s (Gene!) Law, reducing efficiency with increased cost per run. The knee-jerk reaction of throwing more cores at it still doesn’t work even with Sentieon magic.
Optimal resource configuration depends on the goal. If you want fast turnaround and don’t care about the cost then adding more vCPUs works by continuing to make the variant calling and fully paralellizable stages faster. If you want engineering optimization then finding the sweet spot of vCPUs and memory that prevents a CPU efficiency drop is the optimal strategy. If you are concerned with cost and have time to spare then finding the resource specs that gives the lowest cost per run without waiting years is the way to go.
Stay tuned for Part 2 where I compare Sentieon DNAscope to Illumina DRAGEN and NVIDIA Parabricks.
BWA 0.7.19-r1273
Samtools 1.22.1
Picard 3.4.0
SRA Toolkit 3.2.1
bgzip 1.22.1
Sentieon 202503.02
Assembly: GCF_041296265.1
Source: National Center for Biotechnology Information RefSeq
Download
Indexing
SRA accession: SRR6474875
Source: National Center for Biotechnology Information Sequence Read Archive.